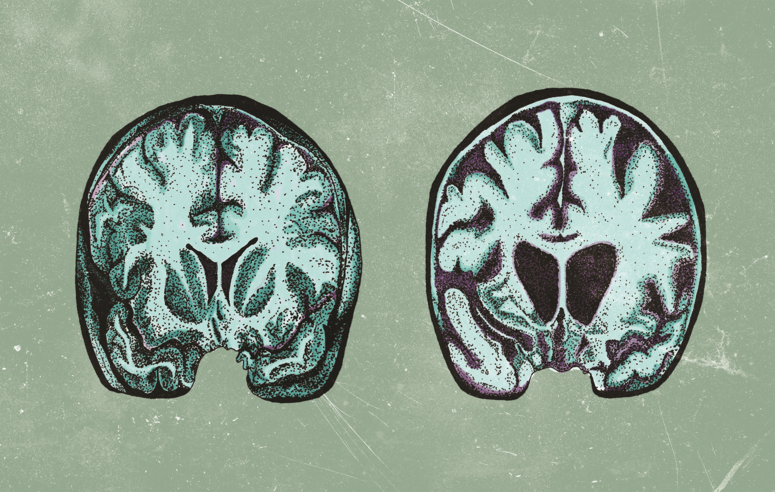
Among inherited neurodegenerative disorders, Huntington’s Disease (HD) is the most common. [1] HD is a grim diagnosis, for the disease not only takes lives but also leaves the descendants of HD patients in fear that they may face the same battle as their parent or grandparent. It’s unsurprising that a diagnosis of HD in the family is cause for distress; in patients affected by HD, a gradual degeneration of neurons leads to major functional deficits. Symptoms include a progressive loss of motor control and cognitive function, as well as the development of psychiatric disorders. [1] Patients with HD commonly begin to experience symptoms around age 40, but the disease can manifest as early as infancy. [2, 3] The first symptoms are often subtle, such as clumsiness, fidgeting, short-term memory loss, and irritability. As the disease progresses, patients struggle to speak or swallow, and they develop involuntary jerking movements known as chorea. Moreover, many experience depression and other psychiatric disorders. [2] The symptoms of HD make it difficult and eventually impossible for patients to carry out daily activities, and their disease ultimately ends their lives within 15 to 20 years of onset. [1] While medications can help manage symptoms, HD is currently incurable. [2] However, research continues to refine our understanding of the disease, and the results of a 2018 study from the Vilchez Lab at the University of Cologne may someday offer a more promising future for HD patients. [4]
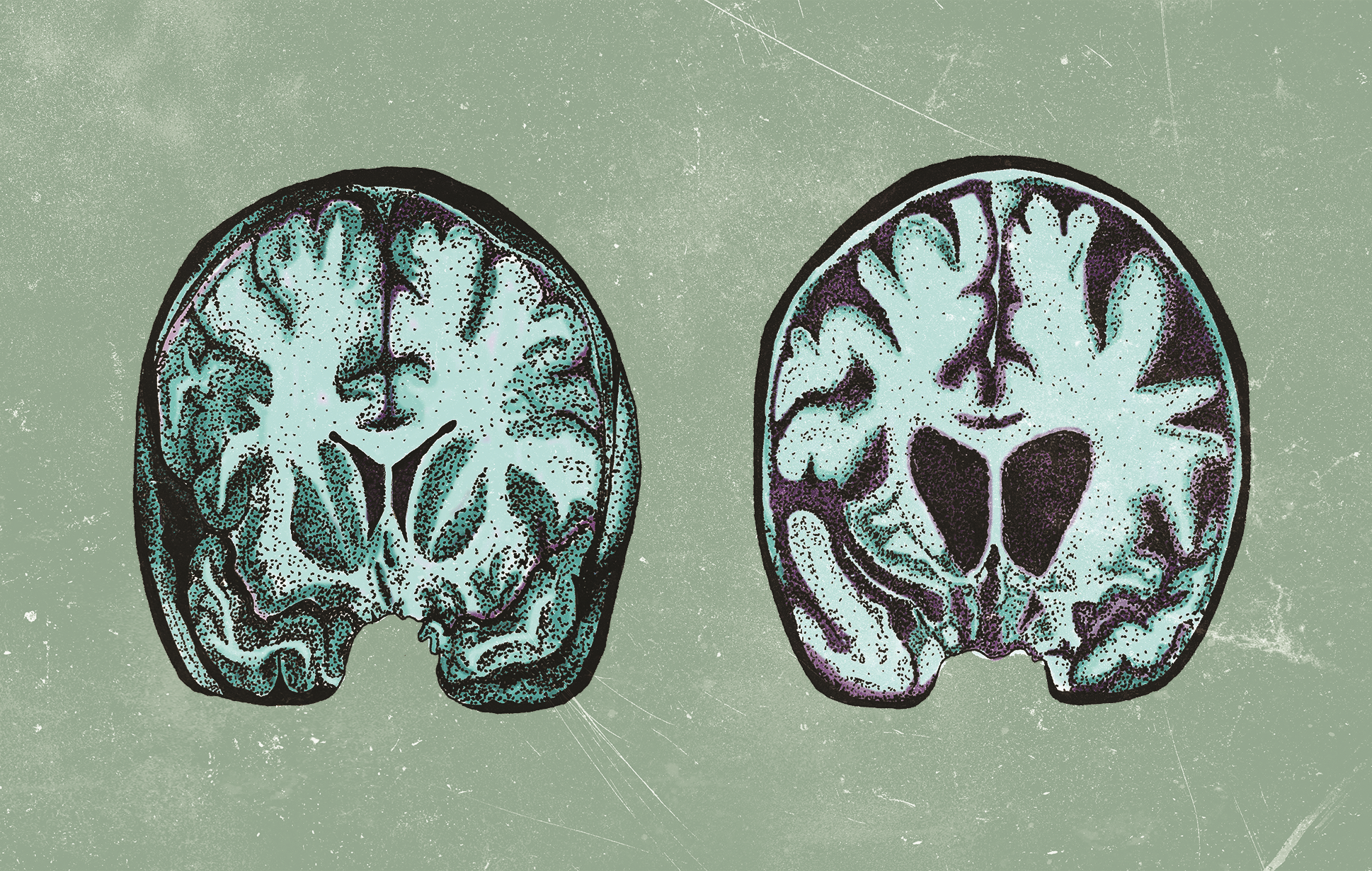
Research surrounding HD often focuses on protein homeostasis, a complex system by which cells regulate protein levels and stability. To understand why protein regulation is of interest to researchers, it is necessary to consider the genetic basis of the disease. DNA consists of a specific sequence of four nitrogenous bases: adenine (A), thymine (T), guanine (G) and cytosine (C), which together form a

blueprint for protein production. HD is an inherited disease caused by a mutation in the huntingtin gene ( HTT ), which codes for the HTT protein. [2] The normal function of HTT is not well characterized, but it is involved in a variety of processes, including transcriptional regulation, apoptosis, and embryonic development. [1] HTT has a segment in which multiple copies of the base sequence “CAG” are present. [5] In healthy individuals, the number of CAG repeats tends to vary from 6 to 35 copies. [4] The mutation responsible for HD results in more than 35 CAG repeats in the gene, leading to an abnormally long HTT protein. As a consequence of its abnormal structure, the mutant protein accumulates and forms aggregates, disrupting neuronal function and causing cell death. [2, 4] Thus, mechanisms for protein regulation and degradation are of great interest to HD researchers.
Stem cells exhibit a unique capacity for enhanced protein regulation, and this essential difference between neurons and stem cells has proved useful in HD research. [4] The enhanced protein regulation in iPSCs is due to tightly regulated mechanisms that prevent accumulation of damaged, misfolded, or aggregated proteins. Induced pluripotent stem cells (iPSCs) are adult body cells that have been reprogrammed to a pluripotent state. This means that, unlike neurons, they can differentiate into many different types of cells. Moreover, while neurons are fully differentiated and do not tend to divide, iPSCs can proliferate indefinitely. Because quality control is critical to cell division, it is essential for iPSCs to maintain an enhanced protein homeostasis network as they divide. For example, the proteasome, a large complex designed to break down abnormal proteins, is very active in iPSCs. Not surprisingly, evidence suggests that HD-affected neurons demonstrate proteasome dysfunction. [4]
Because of this important difference, the Vilchez Lab investigated protein degradation mechanisms in iPSCs derived from HD patients. [4] Previous research has established that like neurons, the iPSCs from HD patients have the characteristic CAG expansion. [6] However, unlike neurons, the iPSCs do not accumulate aggregates of the mutant HTT protein. [4] The researchers hypothesized that this was likely due to the enhanced regulation of proteins in iPSCs, including upregulated proteasome activity. In fact, when the researchers treated these cells with proteasome inhibitors, it triggered an accumulation of HTT. This supported their hypothesis that proteasome activity in iPSCs contributed to the cells’ normal ability to prevent HTT accumulation. [4]
Having confirmed the role of the proteasome in HTT regulation, the researchers then explored the E3 ubiquitin ligase network in iPSCs. E3 ligases catalyze the transfer of a small marker protein called ubiquitin onto other proteins. [4] This ubiquitin tag targets these proteins for degradation by the proteasome. Results indicated that an E3 ligase known as UBR5 was substantially upregulated in iPSCs compared to neurons. Furthermore, when researchers knocked down UBR5 in iPSCs, HTT protein levels increased. Their results link UBR5 to HTT protein modulation and suggest that high expression of UBR5 likely contributes to the increased ability of stem cells to maintain protein homeostasis of HTT in iPSCs compared to neurons. [4] Assuming that accumulation of HTT causes HD, this suggests that developing mechanisms to upregulate UBR5 in neurons could improve HD prognoses.
Although research is far from curing HD, these findings suggest that mimicking the protein homeostasis mechanisms of iPSCs may someday be a plausible treatment for the disease. [4] Moreover, the results identify UBR5 as a possible modulator of HTT protein homeostasis. [4] Future research may further refine the link between UBR5 expression levels and HD, but the current findings are a major step forward in understanding and treating Huntington’s Disease.
References
- Koyuncu, S., Fatima, A., Gutierrez-Garcia, R., & Vilchez, D. (2017). Proteostasis of Huntingtin in Health and Disease. International Journal of Molecular Sciences, 18 (7), 1568. doi:10.3390/ijms18071568
- Saudou, F., & Humbert, S. (2016). The Biology of Huntingtin. Neuron, 89 (5), 910-926. doi:10.1016/j.neuron.2016.02.003
- Walker, F.O. (2007). Huntington’s Disease. The Lancet 369( 9557), 218-228. doi:10.1016/S0140-6736(07)60111-1
- Koyuncu, S., Saez, I., Lee, H. J., Gutierrez-Garcia, R., Pokrzywa, W., Fatima, A., . . . Vilchez, D. (2018). The ubiquitin ligase UBR5 suppresses proteostasis collapse in pluripotent stem cells from Huntington’s disease patients. Nature Communications , 9 (1). doi:10.1038/s41467-018-05320-3
- Macdonald, M. E., Ambrose, C. M., Duyao, M. P., Myers, R. H., Lin, C., Srinidhi, L., . . . Harper, P. S. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell, 72 (6), 971-983. doi:10.1016/0092-8674(93)90585-e
- HD iPSC Consortium (2012). Induced Pluripotent Stem Cells from Patients with Huntingtons Disease Show CAG-Repeat-Expansion-Associated Phenotypes. Cell Stem Cell, 11 (2), 264-278. doi:10.1016/j.stem.2012.04.027