Imagine a human brain. Visualize its characteristic grooves and folds. See how they serpentine across the entirety of its mass, like a thousand rivers through a forest of grey and white matter. Now imagine if all those rivers had been dried up from the start. No more grooves. No more folds. The brain’s surface is now a blank canvas, a mass of cells waiting to be painted with rivers. This blank canvas is the work of a neurological condition called lissencephaly.
What is Lissencephaly?
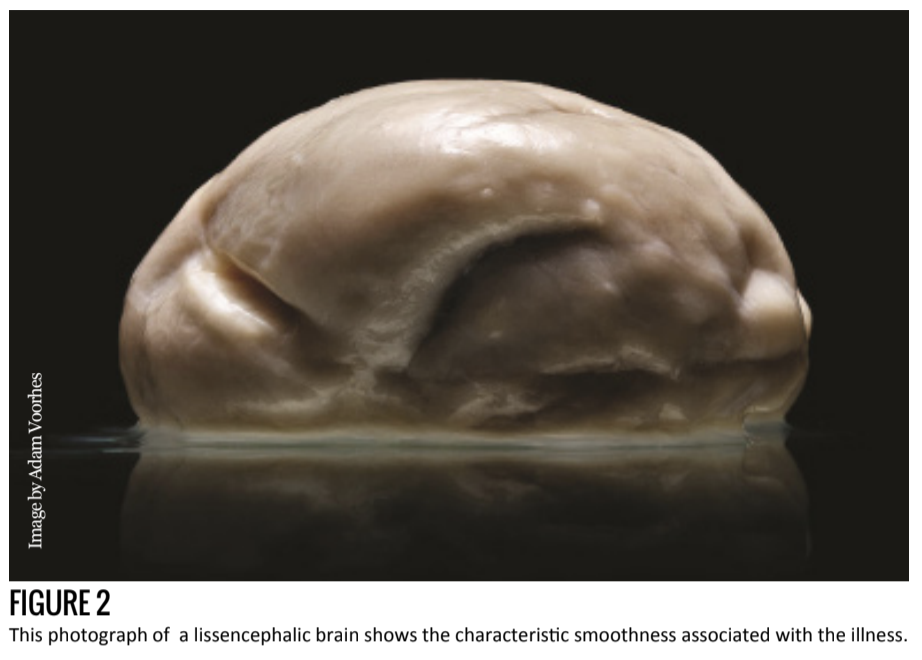
Lissencephaly, literally “smooth brain” from the Greek “lisso” (smooth) and “encephalos” (within the head), is a condition aptly named for its characteristic absence of sulci and gyri. The condition is congenital and has severe - even fatal - consequences for affected individuals. Those who suffer from lissencephaly typically die before the age of ten [1].
Mortality is usually a result of the condition’s symptoms, which include difficulty consuming food or drinking liquid, respiratory diseases, and severe epilepsy. Lissencephaly also hinders intellectual development beyond that of a 3-5 month old child, resulting in mental retardation alongside other developmental challenges [1]. As of April 2013, this severe malformation disorder occurs in 1 of 30,000 births [2].
In terms of physiology, the smoothness of the brain can be attributed to complications in early fetal brain development. As the fetus develops, its cells receive signals from their cellular neighbors to relocate to certain areas in the body for later development and differentiation. Around 12-16 weeks into gestation, specific signaling issues in the brain can result in lissencephaly [3].
In a healthy brain, neuroblasts migrate from deeper regions of the developing brain to specific locations of the cerebral cortex [4]. This process, termed neuronal migration, enables neurons to further differentiate and properly coordinate wiring of synaptic circuits later in development.
As one can imagine, an immense amount of cell signaling and communication is integral to the success of such a process. Migrating neuroblasts have to travel nearly 1,000 cell-body lengths to their specified location in the cortex where they begin to form layers [5]. These cortical layers form radially from the center towards the surface of the brain, meaning that each wave of traveling neuroblasts must maneuver around the layers formed by previous waves of migrated neurons [5]. If this process does not run smoothly, neurons can accumulate near the surface of the cerebral cortex, fail to migrate, or migrate to the wrong locations. Malfunctions in this process are often associated with a variety of genetic mutations that lead to incorrect protein synthesis.

Genetic Mutations
Defective mechanisms can usually be traced back to cell signaling and genetic mutations. In general, a genetic mutation is any permanent change to a sequence of DNA. Two such mutations, which result in lissencephaly, can be due to a permanent deletion of a single DNA building block (known as a nucleotide) or deletion of a rather significant chunk of the chromosome.
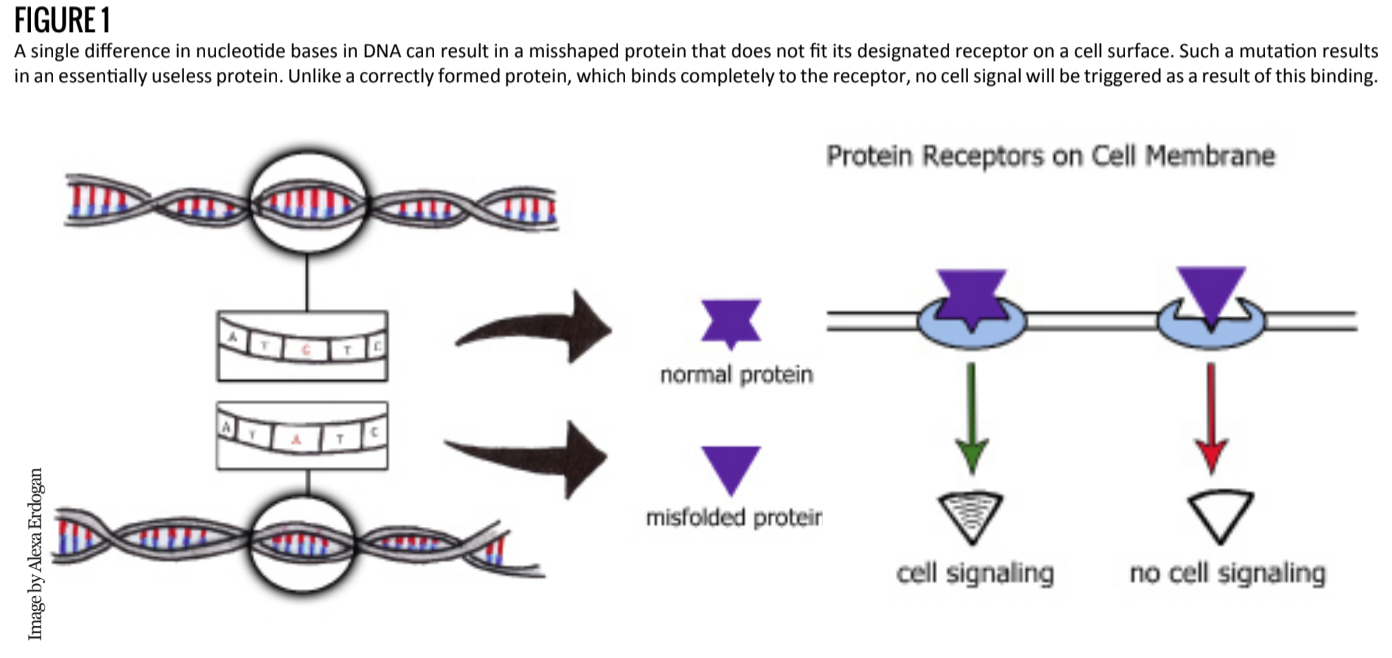
Each gene is like a word, which is comprised of specific letters in a particular order. These individual letters of the alphabet are analogous to individual nucleotides. As an example, the word “neuron” carries significance and can be understood by the reader, but if a letter is deleted, the resulting word, “neron”, makes no sense. Similarly, if several letters are deleted, the reader is left with the term “neon”, which means something entirely different. Since a gene is like the instruction manual for a specific protein, deletions in that gene can cause confusion (neuron vs. neron vs. neon) and prevent a protein from being constructed correctly.
The structure of each protein is integral to its function, and as such, construction errors can lead to partial or complete loss of function (Figure 1). Mutations can be an unfortunate byproduct of DNA replication; one section might not get copied correctly or might be skipped over. While cells do have something akin to “spellcheck” mechanisms to prevent most replication issues, these errors still occur.
In the case of disorders that start at birth, many of these mutations can occur just after fertilization resulting in a new mutation, or a de novo mutation, in that first cell. Every descendent of that cell will then carry that mutated DNA, which can ultimately lead to medical conditions even if there is no family history of that specific genetic mutation.
Mutations Involved in Lissencephaly
Researchers have explored and discovered several genes linked to neuronal development which, when mutated, result in different sub-classifications of lissencephaly. For the sake of brevity, however, this article will focus only on two broad categories: classical lissencephaly (Type I) and cobblestone lissencephaly (Type II).
In classical lissencephaly, improper neuronal migration causes neurons to clump together instead of spreading out and forming the folds and grooves of a healthy brain. As a result of this accumulation, the cerebral cortex becomes abnormally thick (12 -20 mm compared to a typical thickness of around 3-4 mm) [6]. In some reported cases of Type I, patients have also presented with cardiac defects or facial deformations [3].
In addition to abnormal cortical development, defective migration can also result in an absence of cells and consequent lack of development of the corpus callosum. This structure plays a significant role in facilitating the communication between the two hemispheres of the brain by serving as a physical bridge between them. In some cases of lissencephaly, the corpus callosum may be either partially formed or not formed at all, resulting in a bridge with limited or no functionality. By hindering this communication, lissencephaly can result in harmful physical conditions such as lack of coordination, diminished response to external stimuli, and seizures [3][7]. On the genetic level, classical lissencephaly has been associated with mutations on the LIS1, TUBA3/TUBA1A, DCX, ARX, and RELN genes.
Mutations on the LIS1 gene are associated with abnormal neuronal migration. The LIS1 gene regulates a protein that is a subunit of an entire protein complex called platelet activating factor acetyl hydrolase 1B (PAFAH1B). This protein complex regulates the levels of a certain molecule in the brain called platelet activating factor (PAF), which helps direct the movement of developing neuronal cells in the brain. When the LIS1 gene is mutated, the initial protein subunit is abnormally small and rendered nonfunctional. As a result, PAF molecule levels go unregulated, which can hinder cells from migrating to their specified locations or even from migrating at all [8].
The LIS1 gene is also associated with a motor protein called cytoplasmic dynein [9], which is integral to the developing brain in terms of migration and nuclear positioning [10]. Defective dynein proteins can hinder proper neuronal migration and have been associated with numerous other neurodegenerative diseases in addition to lissencephaly [11].
Overall, these issues with neuronal migration can result in several physical abnormalities of the brain, such as a lack of grooves and folds on the brain’s surface. Research has shown that LIS1 mutations are indeed correlated with smoothness of the cortex, particularly towards the posterior end of the brain12. Improper neuronal migration would also cause the formation of larger ventricles, which has been shown using brain imaging techniques [12].
Two other subtypes of classical lissencephaly have been linked to mutations on TUBA1A/TUBA3 and DCX genes. The TUBA1A/TUBA3 gene codes for a protein called alpha-tubulin (α-tubulin), which is involved in the formation and organization of filamentous structures called microtubules [13]. Microtubules are significantly involved in cell structure and movement. One can think of these structures as something akin to the poles comprising the frame of a tent. If certain poles at the top of the frame are bent out of shape, the top of the tent will sag. Similarly, microtubules form a structural frame for the cell (referred to as a cytoskeleton). Defective microtubules can result in a bent frame that hinder the cell’s function and in an inability to properly move developing brain cells to their appropriate locations [14][15][16]. DCX genes, which are located on the X chromosome, regulate a protein called doublecortin, which binds to microtubules and stabilizes them. Doublecortin and microtubules work as a relocation team to help move neurons to their proper locations in the brain. Mutated DCX genes break this team apart by impairing the function of doublecortin, thereby leaving microtubules unstable and disorganized. Without the aid of these two proteins, many developing neuronal cells are thus rendered immobile [17].
Mutations on the ARX gene can also lead to other sub-types of classical lissencephaly. In order to elucidate the function of ARX, researchers developed a test to examine what happens to the brain’s progenitor cells when the gene is inhibited and when the gene is overexpressed. When inhibited, progenitor cells prematurely left the cell-division cycle, thus impairing their eventual migration. On the other hand, overexpression led to an extension of the cell-division cycle, causing cells to multiply uncontrollably. Researchers also tried completely inactivating the ARX gene in which case, cells could no longer develop into the right structure and shape, resulting in limited motility [18]. This evidence suggests that regulated expression of the ARX gene allows cortical progenitor cells to divide and develop in a specific way to guide them towards their cortical destination.
Yet another subtype of lissencephaly has been linked to genetic mutations on a gene called the RELN gene. While the common type of classical lissencephaly is characterized by a strict absence of grooves and folds, RELN mutations actually result in partial but very shallow grooves and folds (a phenomenon called pachygyria) [19]. The RELN gene codes for a protein named reelin. Early in the development of the brain, reelin signals developing neurons to migrate radially outwards in order to form the beginnings of the cortical layers. Later on, reelin promotes the maturation of certain neuronal parts, such as dendrites and dendritic spines. In a mature brain, the protein also plays a role in regulating synaptic function [20]. Naturally, RELN mutations that result in nonfunctional reelin prevent developing neurons from receiving the proper signaling they need in order to migrate correctly. In the grand scheme of things, RELN mutations eventually result in a lack of distinct cortical layer development in the brain.

The second classification of lissencephaly (Type II), also known as cobblestone lissencephaly, is so named for the cobblestone-like appearance of the brain on an MRI scan. In this version of lissencephaly, neuronal development is almost completely disorganized. Consequently, the cerebral cortex suffers a high degree of disorganization and a lack of distinguishable cortical layers. The brain also has a slightly grooved or cobblestone-like surface, which is the result of cortical neurons migrating outwards more than usual. As in the case of Type I, this is due to a defective protein [21].
Type II lissencephaly is further associated with three types of neurological disorders: Walker-Warburg syndrome, Fukuyama syndrome, and Muscle-Eye-Brain (MEB) syndrome [22][23]. However, these three disorders are typically classified as types of congenital muscular dystrophy, thus an exploration into these subtypes would require an in-depth examination of the biological mechanisms behind muscular dystrophy. However, it suffices to state that defective neuronal migration and improper structural formation, similar to those discussed in Type I lissencephaly, also play a part in Type II lissencephaly.
Non Genetic Influences
It should be noted that in addition to genetically linked lissencephaly, there is evidence that suggests there may also be a number of environmental factors involved. Perhaps one of the most significant environmental factors being studied is the introduction of harmful substances or viruses to the fetus during pregnancy.
In 2008, a case study was published that detailed the occurrence of lissencephaly in a fetus with a cytomegalovirus infection (CMV) [24]. CMV is caused by a specific strain of herpes virus and has been known to attack the brain [25] along with other parts of the body. Further research showed that CMV specifically attacks the brain’s cortical progenitor cells during early development [26]. Using mouse models, researchers injected cerebral ventricles with CMV and later observed that this resulted in disrupted neuronal migration as well as a significant loss of neurons [27]. Recently, research has also suggested other contributing environmental factors that include in utero exposure to cocaine [28], ethanol, and ionizing radiation [29].
Conclusion
There remains much to be studied in regards to the causes and treatments of lissencephaly. As with many medical conditions, there are a number of contributing factors in play, meaning there is no singular, simple answer. Scientists are currently focusing on two main angles of attack to better understand the mechanisms behind this disorder.
First, research is being conducted to further explore and elucidate the molecular mechanisms behind specialized and targeted neuronal migration. An enhanced understanding of how neurons receive their signals to migrate to certain areas of the brain can help us identify what goes wrong in these mechanisms.
Second, a deeper analysis of the genes and their mutations might further our understanding of the link between genetic and molecular mechanisms. By comparing mutated proteins to functional proteins, we can better pinpoint the genetic mistakes that contribute to lissencephaly. Utilizing these two research methods can help construct efficient genetic therapies as well as significantly increase our understanding of both the human genome and the brain.