
Our nervous system, generally speaking, is composed of three types of neurons: sensory, motor, and interneurons. Together, these neurons create pathways that transmit signals from all across our bodies via electrical potentials and chemical messages to make us behave the way we do. However, when a neuron cannot function properly, information no longer transmits between it and other neurons. Motor neuron disease is the third most common neurodegenerative disease after Alzheimer’s and Parkinson’s even though it is relatively rare, affecting about 1 in every 50,000 people in the world [1] . The term motor neuron disease classifies a range of diseases that affect the motor neurons, which signal the contraction and movement of muscles within the body [1] . One such disease is amyotrophic lateral sclerosis, or ALS. Stiff muscles, muscle twitching, and weakness due to muscle deterioration characterize ALS, and it can eventually lead to difficulty speaking, swallowing, and breathing [1] . Although improved knowledge of how the nervous system works is refining our understanding of this complex disease, there are still no definitive theories about the causes of ALS. However, increasing experimental evidence points to oxidative stress and glutamate-mediated toxicity as potential causes [2] . There are at least five pathological subtypes of ALS based on the proteins that are affected in each case; here, two are described in detail—TARDBP and SOD1 [3] .
TARDBP Subtype
Recent research has attributed changes within the TARDBP gene to be the primary cause of ALS, which affects the protein TDP-43 [3] . TDP-43 is an RNA/DNA binding protein involved in modifying the structure and composition of RNA (such as removing non-coding portions of RNA). In ALS cases, it is marked for destruction and rendered inactive. When there is no active TDP-43 present, the protein’s structure and function is altered. An important shared feature in most cases of ALS is the accumulation of misfolded proteins. Notably, TDP-43 proteins are major components of protein aggregates found in spinal cord and brain samples of patients with ALS [3] .
Three research groups sequenced the entire coding region of the TARDBP gene and found mutations associated with ALS. Additional research found 47 mutations of TARDBP in cases of ALS that cause a different protein to be formed [3] . Although mutations in TARDBP that affect protein structure and function are the –primary cause of ALS, reactive oxygen species, oxygen molecules capable of eliciting mutation-causing damage which may lead to severe consequences such as dysfunctional protein structures, can also be a cause of ALS.
Oxidative Stress: Reactive Oxygen Species
Oxidative stress occurs when there is a buildup of reactive oxygen species that decrease an organism’s capability for detoxification. Things that contribute to oxidative stress can be as extreme as exposure to radioactive material or as simple as a poor diet [4] .
Previous research has shown that point mutations in the antioxidant enzyme Cu/Zn superoxide dismutase (SOD1) are present in some cases of ALS, most of which are usually inheritable and not necessarily sporadic [4,5] . An antioxidant is a substance that prevents oxidation, the reactive chemical process through which molecules lose electrons. SOD1 is a channel protein and enzyme with copper and zinc binding sites. Normally, it functions as an enzyme that detoxifies copper and zinc, which can enter the cell and are toxic to organelles. It also functions to prevent too much zinc and copper from entering the cell in the first place [4,5] . SOD1 additionally detoxifies reactive oxygen species and prevents these molecules from causing oxidative stress.
Multiple research studies have noted more than 60 different mutations in the SOD1 gene in more than 250 ALS pedigrees [4,5] . They also noticed a lower level of SOD1 activity, as measured in red blood cells, most likely due to the lower stability of the mutated protein [4,5] . These statistics demonstrate that there is a link between mutations in SOD1 and the development of ALS in patients.
When a mutation that causes oxidative stress occurs, a cell attempts to limit or decrease the oxidative stress to which it is subjected [2,6,7] . However, in some cases, these attempts can cause more harm than good to the organism. One such example is that of the protein PARP (poly ADP-ribose polymerase), which is responsible for increasing access to DNA bound to a histone, a protein complex around which DNA coils to form chromosomes, for other repair enzymes. PARP can potentially be very harmful for the cell because it uses large quantities of energy, putting the cell at risk of death [2,6,7] .
Different Hypotheses
Two prevalent theories for how lowered SOD1 activity leads to neuronal dysfunction are the loss of function hypothesis and the alternate hypothesis. The loss of function hypothesis states that toxicity occurs because reactive oxygen species are not detoxified by the mutated SOD1 protein [6,7] . The alternate hypothesis states that the mutant SOD1 protein may be causing motor neuron injury by becoming toxic itself [6] . The mechanism by which SOD1 becomes toxic is proposed to be in the active site of the enzyme, where copper and zinc normally bind. X-ray crystallography, a technique that is used to determine the structure of molecules, has shown that the channel of the mutant protein has a slightly more open configuration than the normal protein. As such, it provides greater accessibility of molecules to the active site. This allows almost any molecule, including reactive oxygen species, to enter the protein channel, accumulate within the protein, and increase its toxicity. This further damages the SOD1 protein [6,7] .
Both hypotheses have been supported in mice studies. Mice carrying several copies of the mutant enzyme developed motor neuron diseases very similar to human motor neuron diseases, while control mice that carried normal SOD1 genes did not [6,7] .

Calcium and Increased Cell Death
Mutated SOD1 proteins are also shown to lead to an increase in cell death via oxidative stress. Under normal conditions, SOD1 protects cells from oxidative stress [6,7] . However, studies showed that injecting mutated SOD1 contributes to an increase in cell death in motor neurons. Researchers proposed that the presence of SOD1 mutations might cause primary defects in a cell’s ability to handle not only reactive oxygen species but also intracellular calcium [6,7] .
Calcium stimulates neurotransmitter release in neurons; therefore, if the calcium levels within the cells are altered, then the amount of neurotransmitters released is altered and by extension the activity of the neuron receiving the neurotransmitter. Researchers found that when mutant SOD1 is expressed, it increases resting cellular calcium concentration. The research group hypothesized that the increase in the calcium concentrations could cause over-excited neural responses compared to the normal responses [6,7] . In some cases, a large influx of calcium, not mediated by SOD1, can cause toxicity and cell death [6,7] . In a study that observed this phenomenon in mice, mitochondria swelled and the dendrites and axons of motor neurons were subject to the formation of vacuoles, which are fluid-containing vesicles within the cytoplasm. Eventually the motor neuron cells form so many vacuoles that they can no longer function and they die [6,7] .
Glutamate and SOD1 Destruction
Research groups noticed that the drastic reduction in the amount of SOD1 proteins present in neural cells could be associated with glutamate receptor proteins. Glutamate is an excitatory neurotransmitter that, once it binds to the receptor of a second neuron, causes an action potential to be fired. Because the concentrations of glutamate decrease, motor neurons no longer fire as many action potentials and over time may become entirely dysfunctional and eventually die [6,7] .
A study observed that in some cases of ALS SOD1 was marked for destruction with a molecular “flag” in all tissues including motor neuron and central nervous system tissues [6,7] . The researchers therefore hypothesized that there must be outside factors besides the mutations within the SOD1 gene that accounts for the degeneration of motor neurons in genetic cases of ALS. One such possible factor could be glutamate. Utilizing different drugs that inhibited glutamate receptors, they noticed that decreased levels of glutamate were toxic to motor neurons. This is most likely because a decreased level of glutamate leads to an increased level of SOD1 proteins tagged for destruction, which decreases the amount of SOD1 proteins available to detoxify reactive oxygen species and other toxic molecules that enter the cell [6,7] .
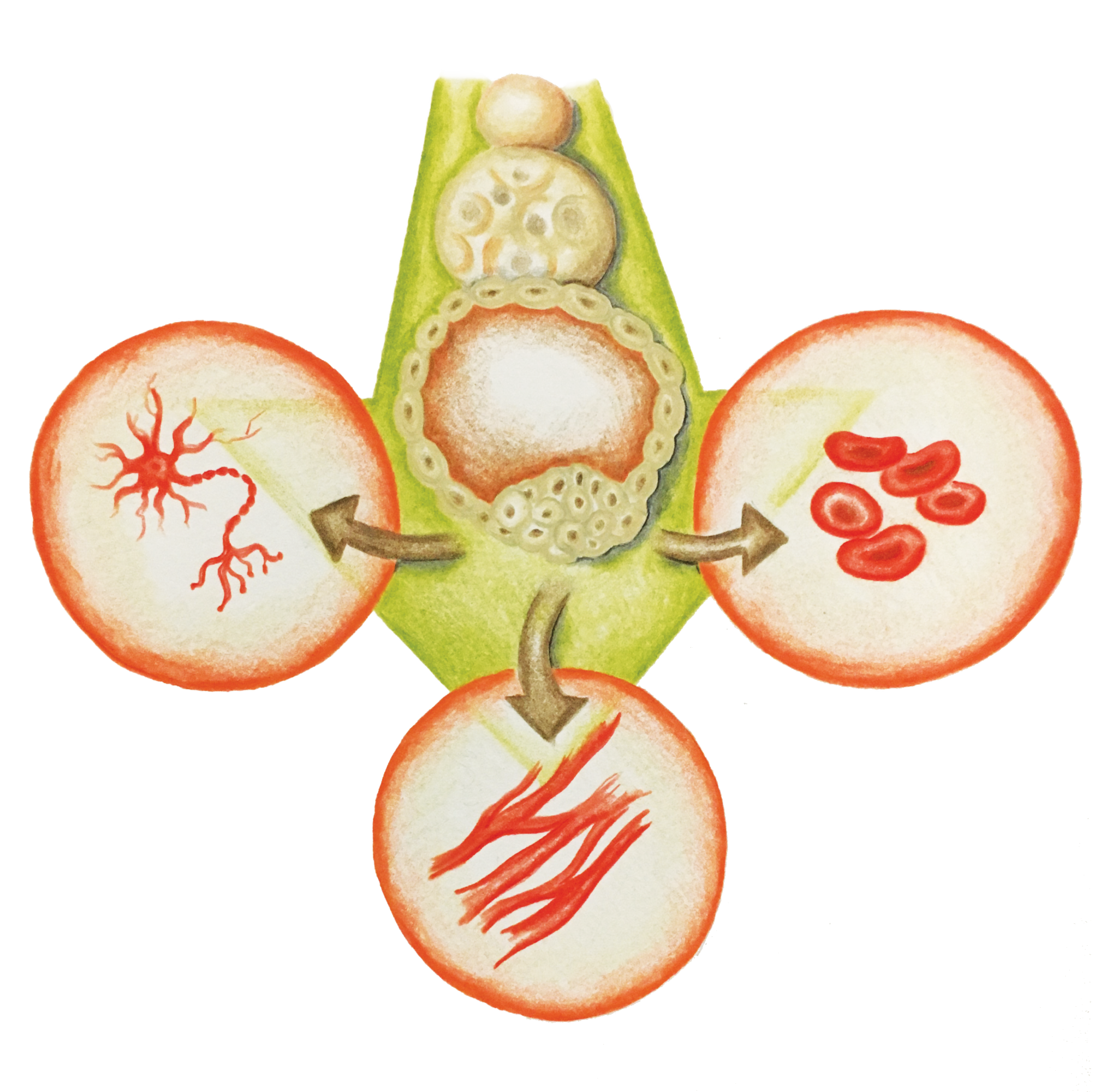
Stem Cells: A New Hope
Recent research has shown that mouse and human fibroblasts (adult, differentiated skin cells) can be reprogrammed to a pluripotent state [8] . A pluripotent state is where a cell can become any differentiated tissue. For example, it can become a heart, liver, brain, or gut cell depending on which proteins that regulate gene expression are expressed in the cell. This means it will behave similarly to an embryonic stem cell. Scientists create pluripotent cells through a process that involves transduction with retroviruses, viruses that, unlike most organisms, transcribe DNA from RNA instead of the other way around. These retroviruses contain the crucial transcription factors for the cell’s differentiation [8] .
A particular study was capable of producing patient-specific motor neurons by using the skin tissue from two siblings, ages 82 and 89, who both possessed the genotype for the SOD1 mutation associated with ALS [8] . The 82-year-old had ALS affecting both her upper and lower body, while the 89-year-old only had upper body motor neuron disease. Two weeks after separating the skin tissue cells and injecting them with the virus containing the transcription factors that enabled the skin cells to become pluripotent, a small number of cell colonies were found with an embryonic stem-cell-like morphology [8] . The researchers were able to use these induced pluripotent stem cells (iPSCs) to create new motor neurons in the two patients. Although complete motor function was not restored in the patients, the transplant was successful and the motor neurons functioned properly when stimulated [8] .
Using embryonic tissue for reprogramming has been met with severe criticism concerning the bioethical, political, technical, and cost issues associated with the therapy [8] . However, iPSCs have the potential for overcoming these obstacles. By using personalized, differentiated adult cells that can be reprogrammed using a simple combination of transcription factors, stem cell therapy for motor neuron disease may soon become affordable enough for wide implementation [8] .
Conclusion
The mechanisms behind the development of ALS are still a mystery. However, our increased understanding of how it progresses in familial situations, such as those with SOD1 mutations, and in sporadic situations, such as those in TDP-43, could improve our knowledge of how to prevent and diagnose early cases of motor neuron disease. In addition, the increasing number of ventures into stem cell regenerative therapy could not only help us devise new methods to treat motor neuron disease in an ethical and affordable manner, but also provide researchers with live models to help study how a variety of motor neuron diseases develop. These promising advancements serve to further enhance our understanding of a complex neurological disease.
- Talbot, K. “Motor Neurone Disease.” Postgraduate Medical Journal. The Fellowship of Postgraduate Medicine, 01 Sept. 2002. Web. 19 Apr. 2017.
- Cookson, Mark R., and Pamela J. Shaw. “Oxidative Stress and Motor Neurone Disease.” Brain Pathology. Blackwell Publishing Ltd, 05 Apr. 2006. Web. 19 Apr. 2017.
- Guererro, Erika N., Haibo Wang, Joy Mitra, Pavana M. Hegde, Sara E. Stowell, Nicole F. Liachko, Brian C. Kraemer, Ralph M. Garutto, K. S. Rao, and Muralidhar L. Hegde. “TDP-43/FUS in Motor Neuron Disease: Complexity and Challenges.” ScienceDirect. Progress in Neurobiology, 28 Sept. 2016.
- Borchelt DR, Lee MK, Slunt HS, Guarnieri M, Xu ZS, Wong PC, Brown RH, Price DL, Sisodia SS, Cleveland DW (1994) Superoxide dismutase-1 with mutations linked to familial amyotrophic lateral sclerosis possesses significant activity. Proc Natl Acad Sci USA 91: 8292-8296.
- Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, Wilcox HM, Flood DG, Beal MF, Brown RH, Scott RW, Snider WD (1996) Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Gen 13: 43-47
- Rothstein JD, Bristol LA, Hosler B, Brown RH, Kuncl RW (1994) Chronic inhibition of superoxide dismutase produces apoptotic death of spinal neurons. Proc Natl Acad Sci USA 91: 4155-4159
- Troy CM, Shelanski M (1994) Down regulation of copper/zinc superoxide dismutase causes apoptotic death in PC12 neuronal cells. Proc Natl Acad Sci USA 91: 6384- 6387
- Dimos, John T., Kit T. Rodolfa, Kathy K. Niakan, Laurin M. Weisenthal, Hiroshi Mitsumoto, Wendy Chung, Gist F. Croft, Genevieve Saphier, Rudy Leibel, Robin Goland, Hynek Wichterle, Christopher E. Henderson, and Kevin Eggan. “Induced Pluripotent Stem Cells Generated from Patients with ALS Can Be Differentiated into Motor Neurons.” Science. American Association for the Advancement of Science, 29 Aug. 2008. Web. 19 Apr. 2017.