
Every day, millions of students experience the feeling of almost indescribable emptiness caused by major depression, also known as major depressive disorder or simply depression. Major depression, one of the most prevalent mental health disorders in the world, is characterized by a loss of pleasure and interest in life. In 2015, approximately 7% of the United States’ adult population experienced at least one major depressive episode within the year [1] . Among college-aged students, major depression episodes had an even higher prevalence, affecting one in every ten individuals [1] .
For a disorder that affects so many people, there is still much left to be understood, as the causes of major depressive disorder are incredibly complex. Accumulated research has revealed that neuronal growth, stress, hormones, and synaptic neurotransmitters—chemicals involved in neuronal communication—all play a role in the manifestation of major depression [2] . Although the growing body of evidence is now creating a much more intricate image of the disorder’s causes, clinical research and treatment tends to emphasize the role of neurotransmitters. The relationship between synaptic neurotransmitter levels and mood was first described by the monoamine hypothesis in the late 1960s [3] . This hypothesis states that a deficiency in monoamine-type neurotransmitters is the biophysiological basis for depression. After observing the unexpected effects of certain medications, psychiatrists hypothesized that depression results from chemical imbalances within the brain. However, new research is revealing just how limited the monoamine hypothesis is. A condition once thought to be caused by an imbalance in chemicals is now proving to be much more complex [2, 3] .
The History and Applications of the Monoamine Hypothesis
Over the course of history, cultures have attributed the causes of major depression to various sources, ranging from unbalanced humors—the four bodily fluids thought to determine one’s health in ancient Greece—to evil spirits. Yet it was a chance medical observation in the early 1950s and the application of the scientific method to this great mystery that brought about one of the most well-known and applied perspectives of the modern era: the monoamine hypothesis [3] .
The formation of the monoamine hypothesis began in 1951, when doctors at the Sea View Hospital on Staten Island decided to administer a new drug to treat patients with tuberculosis. The drug, called iproniazid, was a known inhibitor of enzymes that are responsible for the breakdown of a class of neurotransmitters known as the monoamines. The monoamine neurotransmitter class includes serotonin, norepinephrine, and dopamine [3] . Soon, the staff noticed a significant improvement in the mood of tuberculosis patients who had been withdrawn and depressed for months. This chance discovery of iproniazid’s effects on mood prompted more research that eventually led to the formal creation of the monoamine hypothesis. This also marked the formulation of the first modern biopsychological perspective on depression [3] .
The monoamine hypothesis predicts that a deficiency of monoamine neurotransmitters leads to dysregulations in the mind’s control of mood [4] . This hypothesis revolutionized how healthcare professionals viewed and treated major depression. If major depression was caused by a deficiency in monoamines, then clinicians could treat the condition by simply prescribing a drug that increases these neurotransmitters. Although dopamine and norepinephrine play important roles in the regulation of mood, most of the early research that came out of this hypothesis focused on increasing serotonin [4] .
While many people are unfamiliar with the formal name of the monoamine hypothesis, it has inspired the creation of a publicly recognized class of drugs – the Selective-Serotonin Reuptake Inhibitors (SSRIs). Although iproniazid was taken off the market for causing liver damage, its ability to increase the synaptic concentrations of serotonin inspired further research. Soon, pharmaceutical companies were in hot pursuit of safer, alternative ways to increase serotonin levels [3] . Twenty years later, this research resulted in a second wave of antidepressants entering the market, most of which were of the SSRI family. SSRI drugs, like Prozac and Zoloft, increase serotonin levels by blocking presynaptic transporters, proteins that promote the recycling of serotonin back into the presynaptic neuron. By blocking these presynaptic transporters, SSRIs increase serotonin levels by increasing the amount of time serotonin has to accumulate within the synapse [4] .
Over time, more drug families similar to SSRIs have developed that increase other monoamine neurotransmitters, like norepinephrine and dopamine [3, 4] . For example, several studies have shown the effectiveness of antidepressants that target norepinephrine. In the late 1990s, Dr. Pedro Delgado conducted a series of studies that explored the effects of monoamine depletion and various types of antidepressants on depression [5] . In order to study the effects of serotonin deficiencies, Dr. Delgado and his team decided to alter the diets of their participants so that the experimental group would not consume tryptophan, a necessary ingredient for the body’s synthesis of serotonin that is only acquired through the diet. If we are not able to obtain tryptophan from food, then our serotonin levels will decrease. For patients whose depressive symptoms had responded to SSRIs, more relapsed into depressive episodes during the tryptophan depletion period than those who had responded to antidepressants targeting norepinephrine. The beneficial effects of SSRIs only worked when the body was able to produce serotonin in the first place. Interestingly, patients who had responded to drugs that increase norepinephrine levels experienced a significantly lower relapse rate. This study showed that the mood of people with major depression was directly affected by deficiencies of specific neurotransmitters within the monoamine class. This may imply that deficiencies in different types of monoamine neurotransmitters must be treated in differing ways. In both cases, the majority of major depressive patients improved when taken off of the tryptophan-deficient diet. Dr. Delgado’s studies powerfully supported the monoamine hypothesis, showing how an induced decrease in specific monoamine neurotransmitters can trigger a relapse into major depressive episodes [5] .
The Limitations of the Monoamine Hypothesis
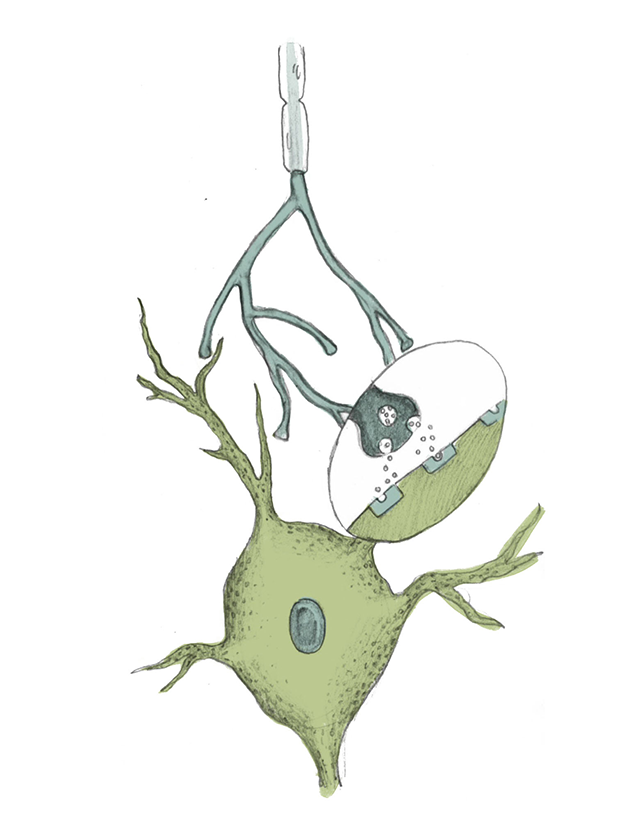
Although the monoamine hypothesis inspired the creation of many drugs which effectively alleviate the symptoms of major depression, it has many limitations. Most notably, SSRIs take several weeks to elevate mood, despite the increase in synaptic monoamine levels after one to three days. Additional questions that seemed to weaken the monoamine hypothesis began to form as further research was conducted at the end of the twentieth century. In a 1994 study conducted at McGill University, male participants with and without a family history of depression were given a chemical mixture that was known to lower synaptic serotonin levels [3] . Over the course of the study, the moods of the participants and their serotonin blood levels were monitored. Interestingly, most participants experienced no change in mood despite a definite decrease in serotonin blood levels [3] . Mood changes were observed only among participants who had a family history of depression. If the monoamine hypothesis were totally true, mood changes would be expected in all participants—not just those with a history of depression. The McGill study would be just one in a long list of studies that began to question whether the monoamine hypothesis alone could accurately explain the biological basis of major depression [3] .
In 2006, more questions were raised after the results of the Sequenced Treatment Alternatives to Relieve Depression (STAR*D study) began to be analyzed [6] . In one of the first federally funded comprehensive studies on the treatment of major depressive disorder, patients across the nation that were diagnosed with moderate to severe major depression were given the option to participate in STAR*D. The STAR*D study consisted of four different levels of treatment. All participants started the study taking the same SSRI (level one). Those who did not experience remission of depressive symptoms went on to try out a new medication or combination of medication and psychotherapy at the second level. Although many types of antidepressants were tried at each level (disregarding level one), 30% of participants who stayed with the study did not experience remission after going through all four levels. The increase of norepinephrine, serotonin, and dopamine by the various drugs could not cure all instances of major depression. The McGill and STAR*D studies both presented the scientific community with results that could not be explained by the monoamine hypothesis alone [3, 6] .
New Perspectives and Research
As more research revealed the missing gaps left by the monoamine hypothesis, more comprehensive hypotheses and theories began to take shape. Within the last twenty years, research in the field of endocrinology—the medical study of glands and hormones—has led to the formation of the neuroendocrine hypotheses for depression. Neuroendocrine hypotheses describe how hormonal responses to stress contribute to major depression [7] . Biological responses to stress are specifically shown to cause hyperactivity in the hypothalamus-pituitary-adrenal (HPA) axis, a system that encompasses many complex hormonal interactions within the body. Changes in such a far-reaching system can have dramatic effects. The slightest increase in activity of the axis can trigger chain reactions that result in large changes in mood. When we encounter a stressor like social rejection or the death of a loved one, our hypothalamus secretes hormones that tell the pituitary gland to release specific directing hormones. These directing hormones are essentially messengers that travel through the bloodstream to our adrenal glands. Once the adrenal glands receive these directing hormones, they begin to secrete the stress hormone cortisol, among other hormones. Together, these three structures play critical roles in the body’s registration of and response to stress [7] .
In 2007, Nicole Vogelzangs and colleagues conducted a study that tested the connection between depression, cortisol, and metabolic syndromes. Vogelzangs defined metabolic syndromes as a cluster of risk factors that increase one’s risk for cardiovascular disease and diabetes [8] . Interestingly, the results showed that depression and high urinary cortisol levels were significantly associated with each other and with metabolic syndromes. Similar hormonal relationships are observed in the mice model of depression [8] . A recent 2017 study by Dr. Shetty and colleagues found that levels of corticosterone, a hormone associated with stress and fear, increased in mice that had been socially isolated [9] . These mice also demonstrated depressive behavior. Many other studies that examine the common relationship of diabetes (a disease that affects the endocrine system) and major depression provide further evidence for the neuroendocrine hypothesis [10] . Although the monoamine hypothesis accurately predicts the changes in neurotransmitter concentrations, it does not predict nor explain the hormonal changes that are also observed in depressed patients [8, 9, 10] .
An accumulation of research in the last fifty years has allowed the psychological community to develop a new theory for understanding the causes of major depression. The neurogenic theory of depression states that an impairment of new neuronal growth (neurogenesis) in the adult hippocampus, a brain structure well known for its role in emotional regulation and memory consolidation, is a biological basis of major depression [11] . In 2013, Dr. Huang and colleagues used Magnetic Resonance Imaging (MRI) to examine the hippocampal volumes of participants with and without major depressive disorder [12] . The study’s results showed a significant difference in the volume of the hippocampus between those with major depression and the healthy controls. Participants with major depression had less tissue in their lower dentate gyrus, a specific fold of hippocampal tissue known for its ability to undergo neurogenesis. Scientists now think this fold of tissue could be the key to understanding the hippocampus’ role in mood disorders. Other studies have shown that a reduction in the size of the hippocampus and an increase in degradation activity (neuron death in the brain) are commonly seen in the post-mortem brains of people who suffered from major depression [11, 12, 13] .
Additionally, many studies show that prolonged usage of effective antidepressants can trigger the birth of new nerve cells and reverse hippocampal shrinkage [14] . This may explain why it takes so long for the mood of patients on antidepressants to improve, despite the rapid increase in monoamine levels. This could also mean that monoamine neurotransmitters play a role early on in the etiological timeline of major depression that culminates in the inhibition of hippocampal neurogenesis. A low serotonin level by itself does not tell the full story of depression. New research is currently exploring neurotransmitters that directly impact neurogenesis (e.g. ceramide). New studies are also showing an association between high cortisol levels and low hippocampal volume, indicating a crossover of the neuroendocrine hypotheses and the neurogenic theory [15] . One of the clearest demonstrations of this crossover was discovered in a 2016 study conducted by Columbia University [16] . In the study, researchers discovered that serotonin receptors on mature mouse neurons were responsible for the various antidepressant effects of SSRIs. An increase in binding to these receptors was shown to trigger other pathways that resulted in the neurogenic and hormonal responses associated with effective antidepressant usage. This new research demonstrates how the monoamine hypothesis interacts with both the neurogenic theory and the neuroendocrine hypotheses to form a clearer picture of the biological basis of major depression [14, 15, 16] .
New Applications in Treatment
As researchers continue to enhance our understanding of the intricate biological mechanisms that underlie major depression, pharmaceutical companies and physicians are taking a more individualized treatment approach. The accumulation of research reveals that there is no one drug type that perfectly “fits all.” New pharmaceutical research is exploring the antidepressant effects of atypical chemical compounds known to increase hippocampal neurogenesis. In a recent 2016 study, Dr. Ma and colleagues at the Shenyang Pharmaceutical University administered a traditional Chinese medicine herbal compound called Xiaochaihutang (XCHT) to mice [17] . Interestingly, mice who had expressed depressive symptoms showed a noticeable change in behavior after receiving XCHT. In addition to an increase in hippocampal volume, depressed mice began to eat more and exhibit less aggressive behavior. By making neurogenesis the end goal of biochemical interventions, pharmaceutical researchers may be able to decrease the time it takes antidepressants to alleviate symptoms. Further studies have also demonstrated the potential for unusual substances, like plant pigments and grape seed extract, to be used as antidepressants. In addition to already in-use SSRIs, these substances have been shown to promote hippocampal neurogenesis and improve mood [18, 19, 20] . The neurogenic theory is inspiring a new frontier in pharmaceutical research that examines a wide range of compounds for potential neuronal growth-promoting and antidepressant properties [17, 18, 19, 20] .
New research is also revealing the effects our daily activities and habits can have on our brain’s ability to regulate emotions and hormones, and generate new neurons. Within the last ten years, a substantial body of research has examined the beneficial effects aerobic exercise can have on major depression and neurogenesis impairment [21] . While aerobic exercise’s ability to alleviate major depression symptoms has been well known for some time, we are still trying to understand the biological mechanisms behind it. New research shows that aerobic activity works to restore adult hippocampal neurogenesis after impairment, leading many healthcare professionals to advise major depression patients to participate in regular aerobic exercise [22] . Additional research in the field of endocrinology also reveals the effects sleep can have on our mood and hormone regulation. Many studies show that sleep deprivation and an increase in wakefulness during sleep increase cortisol levels and the risk of developing major depression [23, 24] . Perhaps the most preventive action we can take against major depression is to prioritize exercise and rest in our daily lives [21-24] .
Although our understanding of major depression has significantly progressed, we are still far from knowing everything about the biological basis of this disorder. However, the future is looking bright. Our biological understanding of major depression has never been this comprehensive. Acknowledging the intricate nature of this disorder will only improve how we treat those who suffer from its symptoms every day.
- Substance Abuse and Mental Health Services Administration. (2015). [Comparative bar graph illustration of SAMHSA data on the prevalence of major depression episodes in the United States in 2015]. 12-month Prevalence of Major Depressive Episode Among U.S. Adults. Retrieved from https://www.nimh.nih.gov/health/statistics/prevalence/major-depression-among-adults.shtml
- Hindmarch, I. (2001). Expanding the horizons of depression: beyond the monoamine hypothesis. Human Psychopharmacology, 16, 203-218. doi: 10.1002/hup.288
- Mukherjee, S. (2012, April 19). Post Prozac nation: the science and history of treating depression. The New York Times. Retrieved from http://www.nytimes.com/2012/04/22/magazine/the-science-and-history-of-treating-depression.html
- Wong, M.L., & Licinio, J. (2004). From monoamines to genomic targets: a paradigm shift for drug discovery in depression. Nature Reviews Drug Discovery, 3, 136-151. doi: 10.1038/nrd1303
- Delgado, P. L. (2000). Depression: the case for a monoamine deficiency. The Journal of Clinical Psychiatry, 61, 7-11.
- (2006). Questions and answers about the NIMH sequenced treatment alternatives to relieve depression (STAR*D) study – all medication levels. National Institute of Mental Health. Retrieved from https://www.nimh.nih.gov/funding/clinical-research/practical/stard/allmedicationlevels.shtml
- Varghese, F. P., & Brown, E. S. (2001). The Hypothalamic-Pituitary-Adrenal Axis in Major Depressive Disorder: A Brief Primer for Primary Care Physicians. Primary Care Companion to The Journal of Clinical Psychiatry, 3(4), 151–155.
- Vogelzangs, N., Suthers, K., Ferrucci, L., Simonsick, E. M., Ble, A., Schrager, M., … Penninx, B. W. (2007). Hypercortisolemic Depression is Associated with the Metabolic Syndrome in Late-Life. Psychoneuroendocrinology, 32(2), 151–159. doi: 10.1016/j.psyneuen.2006.11.009
- Shetty, R.A. & Sadananda, M. (2017). Brief social isolation in the adolescent Wistar-Kyoto rat model of endogenous depression alters corticosterone and regional monoamine concentrations. Neurochemical Research, 1-8. doi: 10.1007/s11064-017-2203-2
- Rustad JK, Musselman DL, Nemeroff CB. (2011). The relationship of depression and diabetes: Pathophysiological and treatment implications. Psychoneuroendocrinology Journal, . 36(9), 1276–1286. doi: 10.1016/j.psyneuen.2011.03.005.
- Miller, B. R., & Hen, R. (2015). The Current State of the Neurogenic Theory of Depression and Anxiety. Current Opinion in Neurobiology, 0, 51–58. doi: 10.1016/j.conb.2014.08.012
- Huang, Y., Coupland, N.J., Lebel, R.M., Carter, R., Seres, P., Wilman, A.H., & Malykhin, N.V. (2013). Structural changes in hippocampal subfields in major depressive disorder: a high-field magnetic resonance imaging study. Biological Psychiatry, 74, 62-68. doi: 10.1016/j.biopsych.2013.01.005
- Sierra, A., Encinas, J. M., & Maletic-Savatic, M. (2011). Adult Human Neurogenesis: From Microscopy to Magnetic Resonance Imaging. Frontiers in Neuroscience, 5, 47. doi: 10.3389/fnins.2011.00047
- Boldrini, M., Butt, T. H., Santiago, A. N., Tamir, H., Dwork, A. J., Rosoklija, G. B., … Mann, J. J. (2014). Benzodiazepines And The Potential Trophic Effect Of Antidepressants On Dentate Gyrus Cells In Mood Disorders. The International Journal of Neuropsychopharmacology / Official Scientific Journal of the Collegium Internationale Neuropsychopharmacologicum (CINP), 17(12), 1923–1933. doi: 10.1017/S1461145714000844
- Moica, T., Grecu, I. G., Moica, S., Grecu, M. G., & Buicu, G. E. (2016). Cortisol and Hippocampal Volume as Predictors of Active Suicidal Behavior in Major Depressive Disorder: Case Report. Balkan Medical Journal, 33(6), 706–708. doi: 10.5152/balkanmedj.2016.150842
- Samuels BA, Anacker C, Hu A, et al. 5-HT1A Receptors on Mature Dentate Gyrus Granule Cells are Critical for the Antidepressant Response. Nature neuroscience. 2015;18(11):1606-1616. doi:10.1038/nn.4116.
- Ma, J., Wu, C.F., Wang, F., Yang, J. Y., Dong, Y.X., Su, G.Y.,… Song, S.J. (2016). Neurological mechanism of xiaochaihutang’s antidepressant-like effects to socially isolated adult rats. Journal of Pharmacy and Pharmacology, 68, 1340-1349. doi: 10.1111/jphp.12616
- Meyer, E., Mori, M.A., Campos, A.C., Andreatini, R., Guimaraes, F.S., Milani, H., & de Oliveira, R.M.W. (2017). Myricitrin induces antidepressant-like effects and facilitates adult neurogenesis in mice. Behavioral Brain Research, 316, 59-65. doi: 10.1016/j.bbr.2016.08.048
- Yoo, D.Y., Kim, W., Yoo, K., Lee, C.H., Choi, J.H., Yoon, Y.S…. Hwang, I.K. (2010). Grape seed extract enhances neurogenesis in the hippocampal dentate gyrus in C57BL/6 mice. Phytotherapy Research Jornal, 25, 668-674. doi: 10.1002/ptr.3319
- Jahromi, M., Razavi, S., Amirpour, N., & Khosravizadeh, Z. (2016). Paroxetine Can Enhance Neurogenesis during Neurogenic Differentiation of Human Adipose-derived Stem Cells. Avicenna Journal of Medical Biotechnology, 8(4), 152–158. doi:
- Alderman, B. L., Olson, R. L., Brush, C. J., & Shors, T. J. (2016). MAP training: combining meditation and aerobic exercise reduces depression and rumination while enhancing synchronized brain activity. Translational Psychiatry, 6(2), e726–. doi: 10.1038/tp.2015.225
- Kempermann, G., Fabel, K., Ehninger, D., Babu, H., Leal-Galicia, P., Garthe, A., & Wolf, S. A. (2010). Why and How Physical Activity Promotes Experience-Induced Brain Plasticity. Frontiers in Neuroscience, 4, 189. doi: 10.3389/fnins.2010.00189
- Matsumoto, Y., Uchimura, N., Ishida, T., Toyomasu, K., Morimatsu, Y., Mori, M., … Ishitake, T. (2016). Day workers suffering from a wider range of sleep problems are more likely to experience suicidality. Sleep and Biological Rhythms, 14(4), 369–376. doi: 10.1007/s41105-016-0067-5.
- An, K. O., Jang, J. Y., & Kim, J. (2015). Sedentary behavior and sleep duration are associated with both stress symptoms and suicidal thoughts in korean adults. The Tohoku Journal of Experimental Medicine, 237, 279-286. doi: 10.1620/tjem.237.279