Neurodegenerative disease is on the mind of the American public. More than five million Americans are living with Alzheimer’s disease, and this number is expected to increase in coming years [1]. Parkinson’s disease has affected a number of prominent celebrities, including Johnny Cash, Muhammad Ali, Michael J. Fox, and most recently, the late Robin Williams [2]. Even awareness and fundraising efforts are at an all-time high – this summer’s social media campaign, the ALS Ice Bucket Challenge, raised $34.2 million towards research for Amyotrophic Lateral Sclerosis (ALS), a neurodegenerative disease also commonly referred to as Lou Gehrig’s disease [3].
Neurodegenerative disease will continue to hold the nation’s attention, as the number of people at risk increases every year. This is because neurodegenerative disease is strongly correlated with age. In the near future, the United States will experience an increasingly large elderly population due to both improvements in healthcare and the aging Baby Boomer population. As a result, we will see a greater number of people affected by neurodegenerative disease. There has never been a more pressing time to understand how these diseases function, develop treatments to slow or halt their progression, and minimize risk factors for our aging population. The first step in solving these complex problems is to elucidate how neurodegenerative diseases operate on a basic cellular level.
Protein Homeostasis: Throwing Off the Balance
One of the most fundamental processes in a cell is maintenance of protein homeostasis by balancing synthesis and degradation. This balance is thrown off in neurodegenerative disease because large protein aggregates accumulate in neurons. For instance, Alzheimer’s disease is marked by the presence of plaques composed of aggregated beta-amyloid and tau proteins [4]. Parkinson’s disease involves the formation of Lewy bodies, which are abnormal protein aggregates that accumulate in the neurons of the substantia nigra pars compacta [4]. Huntington’s disease is a genetic protein aggregation disorder, in which the huntingtin gene encodes an extended glutamine sequence that allows huntingtin proteins to stick together and form aggregates [5]. These protein aggregates are toxic, rendering neurons unable to fulfill their function and ultimately resulting in cell death, or neurodegeneration. While the identification of disease-specific protein aggregates has aided characterization and diagnosis, the mechanism by which protein aggregates promote neurodegeneration remains poorly understood.
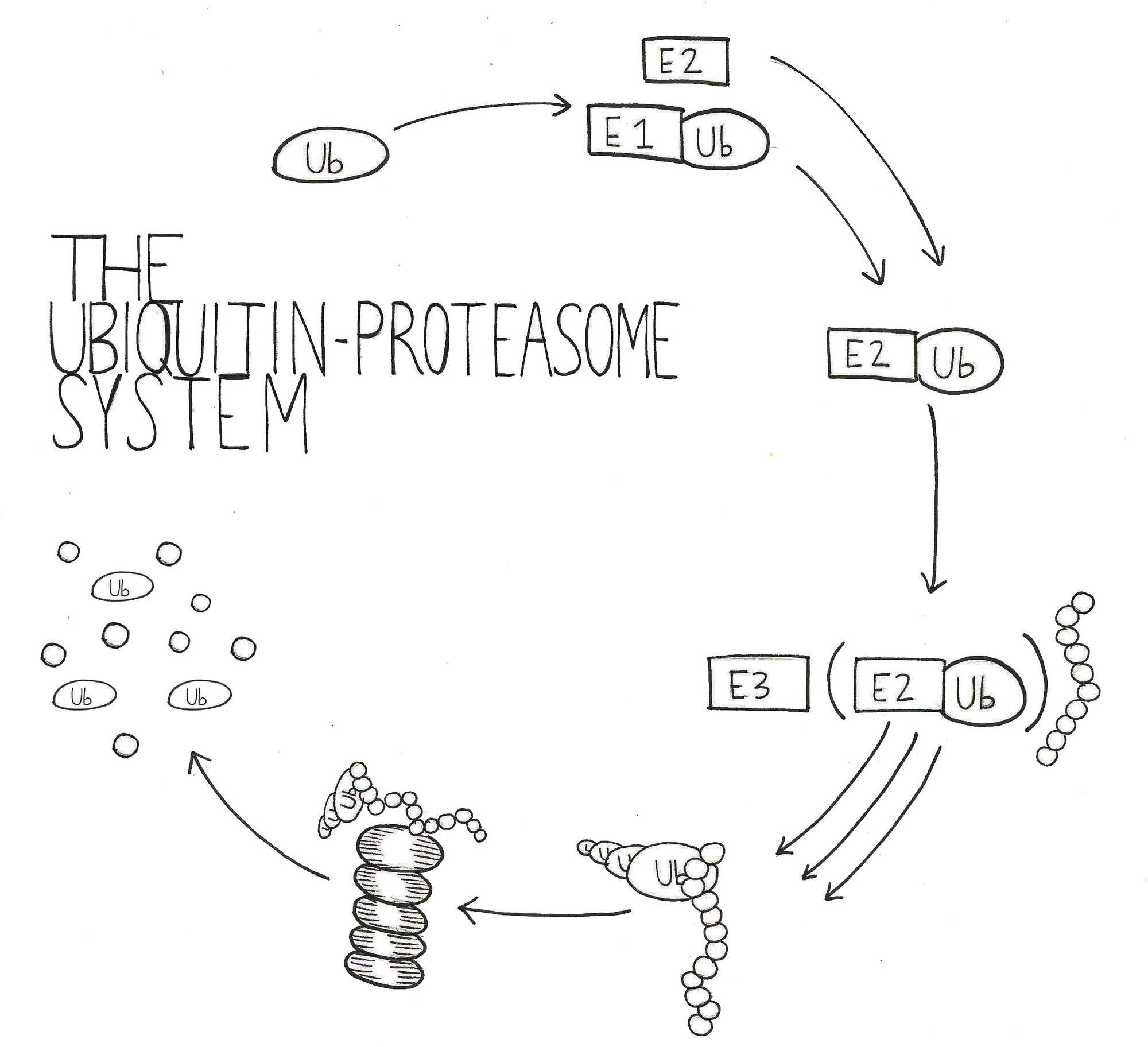
Protein aggregates could be affecting the cell in many different ways that ultimately lead to neurodegeneration. One possible mechanism involves inhibiting the degradation component of the protein homeostasis equation. Degradation is largely accomplished by the ubiquitin-proteasome system (UPS), in which ubiquitin chais are attached to a protein substrate to recruit the proteasome for hydrolysis into short peptide fragments [6]. In a healthy cell, misfolded or unstable proteins are marked for degradation by ubiquitin chains, and targeted to the proteasome for digestion. Additionally, proteins involved in temporary signaling must be rapidly degraded by the UPS to maintain proper cell function. This is especially important in neuronal cells, as signaling by neurotransmitters relies heavily on protein synthesis and turnover. Proteins allow neurotransmitters to be packaged into secretory vesicles and received by protein receptors on other neurons. Indeed, the UPS has been shown to be a key presynaptic regulator that is involved in neurotransmission strength and vesicle formation [7].
However, the presence of large protein aggregates could prevent the UPS from efficiently degrading all of the ubiquitylated proteins within the cell. Decreased UPS efficiency would have harmful impacts on the cell, including diminished presynaptic efficiency, increased levels of misfolded and unstable proteins, and fewer amino acids available for protein synthesis. In order to test the hypothesis that UPS inefficiency is contributing to neuronal toxicity, researchers need to choose an effective model of neurodegenerative disease.
Model Material: Huntington's Disease
Huntington’s disease serves as a good model for neurodegenerative disease because it is monogenic, meaning it is caused by a single gene (the huntingtin gene encoding an extended glutamine repeat) and penetrant, meaning that these genetic effects can be seen in a characteristic phenotype [5]. In contrast, it is more difficult to study Alzheimer’s or Parkinson’s disease because the cause for these diseases remains poorly understood. Rather than being linked to a single gene, Alzheimer’s and Parkinson’s disease are associated with various genes that increase an individual’s risk of developing the disease but are not a direct cause [8][9]. There is also evidence to suggest that environmental factors contribute to disease development, as Alzheimer’s disease may be affected by diet [10] and Parkinson’s disease is associated with head injury and exposure to certain chemicals [11].
Therefore, focusing on Huntington’s disease research provides a simple and effective model for investigating the effect of protein aggregates on the ubiquitin-proteasome system. Furthermore, results from studies in Huntington’s disease are likely to apply to Alzheimer’s and Parkinson’s disease, because these three neurodegenerative diseases share important characteristics such as protein aggregation, neuronal toxicity, and late onset [5]. By studying UPS function in Huntington’s disease models, researchers have gained important insights into how neurodegenerative disease operates on a cellular level.
UPS Impairment
The question of UPS impairment in Huntington’s disease is easier asked than answered. Research on UPS impairment has focused on two hypotheses – proteasome choking and indirect inhibition by changes in the cellular environment. In proteasome choking, large aggregates are thought to get stuck inside the proteasome when being digested into peptides, thus directly inhibiting the proteasome from degrading other proteins. Alternatively, the inhibitory environment hypothesis suggests that the proteasome is capable of degrading protein aggregates, but the presence of aggregates changes cellular conditions in a way that diminishes degradation efficiency.
Direct Impairment: Proteasome Choking
The first indication of UPS impairment in Huntington’s disease was the observation of ubiquitylated huntingtin aggregates, meaning that in the cell, aggregates are tagged for degradation by the proteasome [12]. Ubiquitylation of aggregates directly precedes neurodegeneration, and the aggregates are never fully degraded by the UPS [12]. This observation led to the proteasome-choking hypothesis, where the proteasome is directly inhibited by aggregates clogging its catalytic core. The proteasome is composed of two 19S subunit caps that recognize and remove ubiquitin, unfold the targeted protein, and feed it into the 20S core for degradation. The 20S core’s interior tunnel has catalytic activity that cuts proteins into smaller fragments called peptides, which are degraded further into their amino acid building blocks. Together, these two 19S subunit caps and the 20S core make up the 26S proteasome (subunit addition is not linear).
Early support for the proteasome-choking hypothesis came from a study that showed eukaryotic proteasomes cannot cleave sequences containing a repeated glutamine amino acid (polyQ sequences) [13]. Normally, the proteasome degrades the other sequences of the protein and then releases these polyQ fragments for degradation by different proteases. However, in Huntington’s disease, these sequences are much longer and as a result may fail to exit the 20S core [13]. In this way, extended polyQ sequences would choke the proteasome and directly prevent future protein degradation.
Another way to test the proteasome-choking hypothesis is by isolating proteases from Huntington’s disease tissue and testing their ability to catalyze protein degradation. If the proteasome-choking hypothesis is correct, one would expect to see decreased catalytic activity in proteasomes isolated from Huntington’s disease tissue. However, many studies failed to detect any difference in proteasome activity between diseased and healthy tissue. Proteasomes isolated from a mouse model of Huntington’s disease exhibited equal catalytic activity to healthy mice [14]. Furthermore, knocking out a protein that prevents cleavage of polyQ sequences did not improve proteasome activity, suggesting that the long polyQ sequences in Huntington’s disease are not clogging the 20S proteasome [14].
One study, however, did detect impaired 20S activity in post-mortem human brain tissue of Huntington’s disease patients [15]. It is possible that proteasome choking is occurring in humans and not mice, but this explanation is unlikely due to the study’s failure to control for confounding variables. For instance, measuring catalytic activity in human post-mortem tissue is tricky, as the tissue cannot be preserved as quickly or effectively as in mouse models. In addition, the study did not report total proteasome levels, so diminished activity could be due to fewer proteasomes present in the Huntington’s disease brain tissue than in the healthy brain tissue. Overall, a critical analysis of the study suggests that the observed 20S proteasome inhibition is a reflection of uncontrolled variables and not an indication of physiological significance.
The proteasome-choking hypothesis was further debunked by a study that showed UPS efficiency actually increases in Huntington’s disease [16]. 20S proteasomes were present at equal levels in diseased and healthy tissue, showing that increased activity is not due to upregulation of degradation machinery. Furthermore, two inducible immunoproteasomal subunits, LMP2 and LMP7, were detected in Huntington’s diseased tissue at higher levels compared to healthy tissue [16]. These inducible subunits are known to be expressed in cells mounting an immune response and give the 20S proteasome increased catalytic activity. The presence of LMP2 and LMP7 in Huntington’s disease tissue suggests that the proteasome is not choked by aggregates, but rather that the cell needs enhanced degradation capabilities when aggregates are present. This evidence supports the second hypothesis for UPS impairment in Huntington’s disease: a cellular environment that indirectly inhibits the degradation pathway.
Indirect Impairment: Inhibitory Environment
In light of evidence against direct impairment by proteasome choking, indirect impairment of the UPS could be inhibiting any part of the UPS pathway other than the 20S proteasome. Since the UPS is a system, there are many other components that could be inhibited by the presence of aggregates. For instance, protein aggregates could cause a change in the cellular environment, so molecules that the UPS needs to function (such as ATP) are less available. Additionally, the UPS could simply be overwhelmed – ubiquitylated protein aggregates increase the demand placed on the UPS, which could diminish its capacity to degrade other proteins. These explanations are not mutually exclusive, as a combination of cellular factors could be contributing to UPS impairment in Huntington’s disease. While the inhibited part(s) of the pathway and the mechanism of inhibition have yet to be identified, evidence for indirect inhibition has been published in multiple studies.
The first evidence of indirect inhibition was the observation that unstable proteins accumulate in Huntington’s disease models. Researchers created the unstable protein GFPu as a reporter for UPS function. GFPu is green fluorescent protein (GFP) fused to a degron sequence that signals the protein for degradation by the UPS. GFP allows the protein to be visualized by fluorescence, and the degron sequence ensures that the protein can only accumulate if the UPS is not functioning efficiently. In a cell model of Huntington’s disease, GFPu levels were four times higher in diseased cells than healthy cells [17], showing that unstable proteins accumulate in vitro. Another study observed in vivo accumulation of GFPu in a mouse model of Huntington’s disease [18]. Researchers targeted GFPu to presynaptic and postsynaptic terminals of neurons, and only observed GFPu accumulation when huntingtin aggregates were present [18]. These results indicated that UPS inhibition is occurring in critical signaling regions of neuronal cells.
In contrast, a third study failed to observe elevated GFPu levels in mouse models of spinocerebellar ataxia, which is similar to Huntington’s disease in that polyQ protein aggregates cause neurodegeneration. However, neurodegenerative symptoms in spinocerebellar ataxia are different than in Huntington’s disease, and include ataxia (loss of muscle control), retinal cell death, and brainstem dysfunction [19]. In this study, GFPu levels were not elevated in retinal tissue in diseased vs. healthy mouse retinas, indicating that UPS impairment does not contribute to the progression of this neurodegenerative disease [19]. Since these results conflict with studies in models of Huntington’s disease, it is important to consider the tissue-specific factors that may be producing these differences. Brain tissue and retinal tissue are uniquely differentiated, so different cellular environments in each tissue type may be responsible for these conflicting results. In addition, a characteristic trait of spinocerebellar ataxia is differential regulation of photoreceptor genes – some genes are upregulated in the retina to increase expression [19]. After examining the reporter gene GFPu, the study found that its expression was upregulated in diseased retinal tissue, meaning more mRNA was available to be translated into protein [19]. An ideal reporter would have equal levels of expression in the diseased and healthy tissue, so that protein levels are not confounded by different levels of transcription and translation. Due to these two experimental problems, it is unlikely that these results would invalidate the hypothesis of indirect UPS impairment.
Indirect UPS impairment has been further supported in experiments using other methods of measurement. For instance, one study used mass spectrometry to get a big-picture assessment of the total level of ubiquitylated proteins within Huntington’s disease brain tissue. Importantly, the study observed proteins ubiquitylated by a K48 linkage, which destines proteins for degradation by the proteosome. Since other ubiquitin linkages have diverse cellular roles, K48 linkage specifically must be examined to analyze UPS function. If more K48 ubiquitylated proteins are present in Huntington’s disease brain tissue, that would indicate that the UPS cannot efficiently degrade proteins in the presence of huntingtin aggregates. By analyzing two different mouse models and brain tissue from human Huntington’s disease patients, this study found that levels of K48 ubiquitylated proteins were in fact elevated in comparison to healthy brain tissue [20]. Higher levels of K48-linked ubiquitin chains further confirmed UPS impairment by an indirect mechanism.
A large body of evidence has supported that indirect impairment of the UPS is indeed happening in Huntington’s disease, and some studies have begun to address the mechanism by which impairment could occur. So far, two mechanisms have been proposed. First, aggregates may overwhelm the UPS by contributing to higher levels of ubiquitylated proteins that UPS cannot efficiently degrade. Second, protein aggregates may cause a change in the cellular environment that slows the UPS’s rate of degradation.
There is evidence to suggest that both of these scenarios are happening, and they can combine to create an inhibitory environment that diminishes UPS efficiency. The first scenario, that aggregates overwhelm the UPS by contributing to higher levels of ubiquitylation, was supported by a study that observed a “critical threshold” of aggregate concentration needed to decrease UPS efficiency [21]. This study also found that overwhelming the UPS did not occur due to proteasome choking, but rather by competition for binding to the 19S proteasome cap [21]. This observation is supported by research showing that huntingtin aggregates directly bind to the 19S cap [22]. Together, these studies support competition for proteasome binding as a mechanism by which aggregates overwhelm and impair the UPS.
The second scenario, that protein aggregates cause an inhibitory change in the cellular environment, is supported by research on mitochondrial function in Huntington’s disease. Altered mitochondrial activity may be creating an inhibitory cellular environment by changing the synthesis and release of molecules that affect UPS function. For instance, huntingtin aggregates associate with the mitochondria in mouse brain models, and this association interferes with mitochondrial transport and ATP production [23]. Since the 19S cap requires ATP to unfold proteins and feed them into the 20S core, diminished ATP levels would hinder degradation efficiency. In turn, the diminished degradation capacity of the UPS affects the synthesis and secretion of molecules from the mitochondria, leading to neuronal toxicity and subsequent death. Specifically, one study found that the UPS’s failure to degrade the cell-cycle protein p53 triggered the release of apoptosis proteins such as caspases and cytochrome c [24]. Therefore, UPS inefficiency may actually trigger the mitochondria to promote cell death in neurons. These two studies suggest a dual interaction between the UPS and the mitochondria, as the UPS may be impaired in response to mitochondrial activity, and in turn, the mitochondria may respond to UPS impairment by promoting neurodegeneration.
Therapeutic Implications
While the exact mechanism of UPS impairment in neurodegenerative diseases remains to be elucidated, it has become clear that UPS function is impaired in Huntington’s disease by an inhibitory cellular environment. This information can be used to propose therapeutic targets that could slow or even halt the progression of neurodegenerative disease. One promising strategy is to use UPS stimulatory drugs to combat the toxic effects of degradation impairment. Researchers have already seen lower levels of neurodegeneration in Huntington’s disease mouse models treated with UPS stimulators baclofen [25] and sulforaphane [26].
While a UPS stimulatory drug has not yet been tested in humans, UPS repressor drugs are already being prescribed to cancer patients. Interestingly, 36% of cancer patients who take UPS repressor drugs develop neurodegenerative symptoms [27], suggesting that UPS stimulators may in fact have neuroprotective properties.
Aside from UPS stimulators, patients suffering from neurodegenerative disease may also benefit from drugs preventing the formation of protein aggregates, as fewer aggregates would lessen UPS impairment. For instance, upregulating chaperones that direct proper protein folding and enhancing disaggregation pathways would decrease the potency of huntingtin aggregates in UPS impairment [28]. If UPS impairment proves to be a difficult problem to solve, patients could also benefit from drugs stimulating other protein degradation pathways outside of the UPS, including autophagy. While autophagy is a less common pathway by which cells degrade proteins, previous research has shown that it is possible for a cell to degrade huntingtin aggregates through autophagy [29]. Autophagy may also help clear other proteins destined for degradation in order to minimize the toxic effect of UPS impairment in neurodegenerative disease. Research identifying drugs that can accomplish these cellular roles, as well as continued research towards understanding the mechanisms underlying neurodegenerative disease, will bring us closer to improving the lives of the millions of people living with Huntington’s, Parkinson’s and Alzheimer’s disease.
References
- http://www.niehs.nih.gov/research/supported/diseases/neurodegenerative/
- http://www.theparkinsonhub.com/be-inspired/celebrity-spotlight.html
- http://www.alsa.org/news/media/press-releases/ibc-initial-commitment.html
- Irvine, G.B., El-Agnaf, O.M., Shankar, G.M., and D.M. Walsh. Protein aggregation in the brain: the molecular basis for Alzheimer’s and Parkinson’s diseases. Mol Med. 14: 451-464 (2008).
- Ross, C. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 10: 83-98 (2011).
- Kleiger, G. and T. Mayor. Perilous journey: a tour of the ubiquitin–proteasome system. Trends in Cell Biology. 24: 352-359 (2014).
- Speese, S.D., Trotta, N., Rodesch, C.K., Aravamudan, B., and K. Broadie. The ubiquitin proteasome system acutely regulates presynaptic protein turnover and synaptic efficacy. Curr Biol. 13: 899 – 910 (2003).
- Chouraki, V. and S. Seshadri. Genetics of Alzheimer’s disease. Adv Genet. 87: 245-294 (2014).
- Chorfa, A., Lazizzera, C., Bétemps, D., Morignat, E., Dussurgey, S., Andrieu, T. and T. Baron. A variety of pesticides trigger in vitro α-synuclein accumulation, a key event in Parkinson's disease. Arch Toxicol. (2014).
- Mosconi, L., Murray, J., Tsui, W.H., Li, Y., Davies, M., Williams, S., Pirraglia, E., Spector, N., Osorio, R.S., Glodzik, L., McHugh, P. and M.J. de Leon. Mediterranean diet and magnetic resonance imaging-assessed brain atrophy in cognitively normal individuals at risk for Alzheimer's disease. NIH-PA Author Manuscript. 1: 23-32 (2014).
- Sundman, M., Hall, E. and N. Chen. Examining the relationship between head trauma and neurodegenerative disease: A review of epidemiology, pathology and neuroimaging techniques. NIH-PA Author Manuscript. (2014).
- Davies, S., Turmaine, M., Cozens, B., DiFiglia, M., Sharp, A., Ross, C., Scherzinger, E., Wanker, E., Mangiarini, L., and G. Bates. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 90: 537-548 (1997).
- Venkatraman, P., Wetzel, R., Tanaka, M., Nukina, N., and A. Goldberg. Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol Cell. 14: 95-104 (2009).
- Bett, J.S., Goellner, G.M., Woodman, B., Pratt, G., Rechsteiner, M. and G.P. Bates. Proteasome impairment does not contribute to pathogenesis in R6/2 Huntington’s disease mice: exclusion of proteasome activator REG as a therapeutic target. Hum Mol Genet. 15: 33-44 (2005).
- Seo, H., Sonntag, K., and O. Iacson. Generalized brain and skin proteasome inhibition in Huntington’s disease. Ann Neurol. 56: 319-328 (2004).
- Diaz-Hernandez, M., Hernandez, F., Martin-Aparicio, E., Gomez-Ramos, P., Moran, M., Castano, J., Ferrer, I., Avila, J., and J. Lucas. Neuronal induction of the immunoproteasome in Huntington’s disease. Journal of Neuroscience. 23: 11653-11661 (2003).
- Bence, N.F., Sampat, R.M., and R.R. Kopito. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 292: 1552-1555 (2001).
- Wang, J., Wang, C.E., Orr, A., Impaired ubiquitin – proteasome system activity in the synapses of Huntington ’ s disease mice. Journal of Cell Biology. 180: 1177-1189 (2008).
- Bowman, A., Yoo, S.Y., Dantuma, N.P., and H.Y. Zoghbi. Neuronal dysfunction in a polyglutamine disease model occurs in the absence of ubiquitin–proteasome system impairment and inversely correlates with the degree of nuclear inclusion formation. Hum Mol Genet. 14: 679-691 (2005).
- Bennett, E.J., Shaler, T.A., Woodman, B., Ryu, K.Y., Zaitseva, T.S., Becker, C.H., Bates, G.P., Schulman, H., and R.R. Kopito. Global changes to the ubiquitin system in Huntington’s disease. Nature. 448: 704-709 (2007).
- Hipp, M.S., Patel, C.N., Bersuker, K., Riley, B.E., Kaiser, S.E., Shaler, T.A., Brandeis, M., and R.R. Kopito. Indirect inhibition of 26S proteasome activity in a cellular model of Huntington’s disease. Journal of Cell Biology. 196: 573-587 (2012).
- Diaz-Hernandez, M., Valera, A.G., Moran, M.A., Gomez-Ramos, P., Alvarez-Castelao, B., Castano, J.G., Hernandez, F., and J.J. Lucas. Inhibition of 26S proteasome activity by huntingtin filaments but not inclusion bodies isolated from mouse and human brain. Journal of Neurochemistry. 98: 1585-1596 (2006).
- Orr, A.L., Li, S., Wang, C.E., Li, H., Wang, J., Rong, J., Xu, X., Mastroberardino, P.G., Greenamyre, T., and S.J. Li. N-Terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. NIH-PA Author Manuscript. 28: 2783-2792 (2008).
- Jana, N.R., Zemskov, E.A., Wang, G., and N. Nukina. Altered proteasomal function due to the expression of polyglutamine-expanded truncated N-terminal huntingtin induces apoptosis by caspase activation through mitochondrial cytochrome c release. Hum Mol Genet. 10: 1049-1059 (2001).
- Kim, W. and H. Seo. Baclofen, a GABAB receptor agonist, enhances ubiquitin-proteasome system functioning and neuronal survival in Huntington’s disease model mice. Biochemical and Biophysical Research Communications. 443: 706-711 (2014).
- Liu, Y., Hettinger, C.L., Zhang, D., Rezvani, K., Wang, X., and H. Wang. Sulforaphane enhances proteasomal and autophagic activities in mice as a potential therapeutic reagent for Huntington’s disease. Journal of Neurochemistry. 129: 539-547 (2014).
- Richardson, P., Mitsiades, C., Hideshima, T., and K.C. Anderson. Bortezomib: Proteasome inhibition as an effective anticancer therapy. Annu Rev Med. 57: 33-47 (2006).
- Balch, W.E., Morimoto, R.I., Dillin, A., and J.W. Kelly. Adapting proteastasis for disease intervention. Science. 319: 916-919 (2008).
- Sarkar, S. and D.C. Rubinsztein. Huntington’s disease: degradation of mutant huntingtin by autophagy. FEBS Journal. 275: 4263-4270 (2008).