
Finding New Ways of Thinking About Depression
Within the last decade, a new agent has appeared in the spotlight of psychiatric research: ketamine. This drug, also known as “Calypsol,” is currently used as anesthesia [1]. Intriguingly, scientists have found that it may alleviate symptoms of major depressive disorder (MDD) when given in low, subanesthetic doses. Many clinical studies have verified that its antidepressant effect can be observed within 24 hours and the effect of one dose may last up to 14 days, which can be prolonged by repeated injections [2]. This makes ketamine potentially more effective than current antidepressants, which do not have the same fast-acting and enduring effects, although ketamine’s long-term potential still needs to be studied carefully.
A patient experiencing MDD suffers from a long-lasting depressed mood and a decreased feeling of pleasure [3]. For a long time, researchers thought MDD was connected to reduced activity of neurotransmitters of the monoamine family. This elicited the development of medication that focused primarily on increasing the levels of these monoamine neurotransmitters, especially serotonin. One class of drugs designed for this purpose is the selective serotonin reuptake inhibitors (SSRIs), which are the current standard therapy against depression. However, as we explored in Issue 10, the monoamine hypothesis was put into question by the results of a large study conducted in 2006, called the “Sequences Treatment Alternatives to Relieve Depression (STAR*D) study.” This study found that fewer than 30% of the almost 3,000 cases of severe MDD that were selected from all over the United States experienced remission after the standard SSRI therapy [4]. Current medications take a long time to become effective and are not suitable for every patient. This suggests that there must be other neurological systems involved in MDD. The astonishing discovery that ketamine, a drug that involves glutamate rather than monoamines, has rapid-onset antidepressant effects even in patients that are resistant to classic antidepressants opened the path for different hypotheses [5][6]. New approaches do not focus on monoamines but on glutamate signaling, the involvement of stress and the hormonal system, and neuroplasticity.
Why We Need a Glutamate-Hypothesis for Depression
So far, there is no approved antidepressant targeting the glutamatergic system directly, but this is likely to change, as a great num-ber of studies are now focusing on the antidepressant potential of ketamine and similar agents. Glutamate is the brain’s major excitatory neurotransmitter and is associated with cognition, emotion, and neuroplasticity. It particularly modifies the strength of synaptic connections and promotes synaptic growth. This plays a large role in everyday neural functions – like memory, for example. Its activity is counterbalanced by GABA, the major inhibitory neurotransmitter. Other neurotransmitters, such as serotonin, modify the basic excitatory and inhibitory signaling provided by glutamate and GABA. They make up a much smaller quantity of neurons in more specific brain areas, and yet, researchers have spent much more energy in investigating their role in depression than looking at glutamate and GABA [5] .
Only recently has the attention shifted towards a new
hypothesis about a GABA/glutamate imbalance in neurodegenerative and psychiatric disorders, including MDD. Indeed, there is evidence that the glutamatergic circuitry contributes to the onset of depression. Studies confirm its dysregulation in rodent MDD models and in human patients, and suggest that this is associated with abnormal structural and functional changes of the excitatory circuitry in the brain [5, 7] . In particular, the plasma, cerebrospinal fluid, and brain tissue of MDD patients all reveal elevated glutamate levels in both in vivo and post mortem studies [5] .
Ketamine is a Nonselective NMDA Antagonist
Scientists have slowly begun to uncover the molecular mechanisms initiated by ketamine, which allows us to understand how it affects the glutamate system and what causes its antidepressant effects. Ketamine disrupts normal glutamate signaling by blocking a specific kind of glutamate receptor in the synapse, called the NMDA receptor (NMDA-R) [2, 5, 6, 7, 10] . NMDA-Rs are widely distributed in the nervous system, yet they cannot respond to a glutamate signal right away since they are blocked by a magnesium ion.
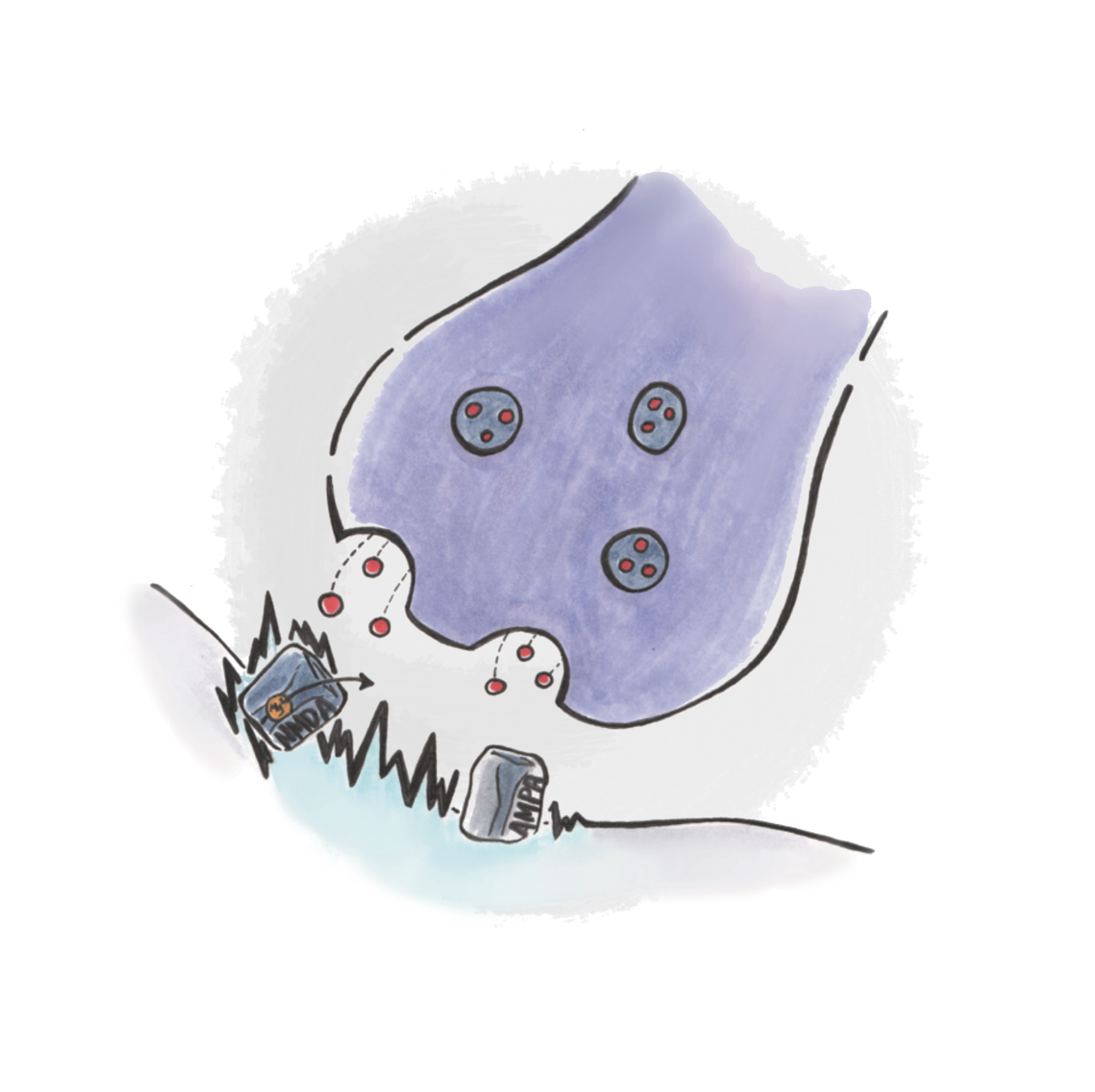
A different type of glutamate receptor, called the AMPA receptor (AMPA-R) canhelp to remove the magnesium “plug” that prevents the NMDA-Rs from signaling. When a glutamate molecule binds to an AMPA-R, it causes its channel to open and allow positively charged ions to enter the neuron. The inside of a cell normally has a slightly negative charge compared to its surroundings, but by adding positive charge, the cell depolarizes, making its inner electrical potential more positive. This condition allows the NMDA-R to “unplug,” as the electrical force now repels the positively charged magnesium ion. The NMDA-R can then respond to stimulation by glutamate and allow calcium ions to enter the cell. Calcium ions are second messengers that can induce a variety of molecular pathways that lead to synaptic remodeling [8] . By blocking, or “antagonizing,” this receptor channel, ketamine prevents the flow of calcium ions and leads to changes in synaptic remodeling.
Ketamine Modifies Molecular Pathways to Induce Synaptic Growth
It seems paradoxical that ketamine, which competitively binds to the NMDA-Rs and therefore increases glutamate in the synapse, decreases the symptoms of MDD, a condition that is also correlated with increased amounts of glutamate. Frankly, the underlying mechanism is poorly understood, and researchers are still investigating this counterintuitive effect. An early model proposes that the blockage of NMDA-Rs causes neurons to release more glutamate, which would then increasingly activate AMPA and other non-NMDA receptors [2] . Newer studies hypothesize a more complex mechanism involving the disinhibition of inhibitory GABA-interneurons, which, under the influence of active NMDA-Rs, would normally downregulate the glutamate signal. As ketamine prevents the activation of NMDA-Rs, and thus prevents the regulatory effect of GABA-interneurons, it causes a burst of glutamate, which activates AMPA-Rs [6] .
AMPA-activation stimulates the formation of new synapses by increasing the expression of a protein known as brain-derived neurotrophic factor (BDNF) [2, 5, 6] . Elevated BDNF after ketamine administration has been observed in the hippocampus and the prefrontal cortex (PFC) [5, 7, 9, 10] . While the hippocampus is a brain structure involved in memory, the PFC refers to the foremost part of the frontal lobe, where complex cognitive tasks, including decision-making and regulation of emotions, are performed. In fact, these two structures have been shown to be smaller in a subset of depressed patients, supporting the hypothesis that MDD is related to decreased BDNF signaling in the hippocampus and in specific areas of the forebrain [6, 10] . Thus, low levels of BDNF in the hippocampus seem to lead to symptoms of depression, while increasing BDNF in the hippocampus should reverse them. In a direct approach, scientists were able to replicate the effects of ketamine by infusing the hippocampus with BDNF; the administration of BDNF induced antidepressant effects, while its genetic knockout control produced the contrary [10] .
Ketamine induces overexpression of BDNF, which rapidly activates the mTOR pathway. This pathway is a chain of proteins that interact with each other to stimulate the synthesis of synaptic proteins for the formation, maturation, and functioning of new synapses [6, 11] . Li et al. studied the mTOR pathway in the PFC of rats and found that ketamine activates it in a dose dependent manner; mTOR signaling is active in a range between 5 to 10 mm/kg of ketamine, but not when administered at the higher anesthetic dose. A single, low dose of ketamine increased the amount of synaptic proteins and the formation of new synapses, which induced stronger connections and higher excitability [11] .
Furthermore, the scientists tested whether mTOR was necessary for ketamine’s antidepressant effects by injecting rapamycin, an agent that blocks the mTOR-pathway. This resulted in the lack of antidepressant effects, suggesting that this pathway is a necessary mechanism that causes ketamine to be effective in a rapid and sustained manner [11] . Taken together, ketamine modulates the amount of BDNF, leading to increased activation of the mTOR pathway. This pathway eventually increases the expression of synaptic proteins that strengthen synapses in a region-specific way, reducing symptoms of depression. Notably, ketamine not only increased responsiveness to glutamate-induced excitatory signals, but also to serotonin, which indicates that ketamine also affects the “classical” monoamine pathways involved in MDD [11] .
Ketamine Increases Stress-Resilience
While reduced levels of BDNF are observed in the hippocampus and PFC of depressed patients, the hypothesis that increasing BDNF would relieve symptoms is a little too simplistic. Further studies have revealed that in other brain structures, especially those involved in the dopamine (a neurotransmitter involved in reward) circuitry, increasing levels of BDNF exerted the opposite effect. Infusions of BDNF into the ventral tegmental area (VTA) and nucleus accumbens (NAc), which are both structures of the dopamine circuitry, were shown to increase, rather than decrease, depressive-like behavior in rodents [10, 12] . Stress is considered to affect the dopamine system in a similar way, underscoring the notion that stressful life events contribute to the onset of MDD [12] .

Although there is no genetic knockout mutant line for depression, its symptoms can be modulated in mice by either exposing them to extreme environmental stress or repeatedly injecting stress hormones [10, 12] . Both models are commonly used in depression studies and confirm the role of environmental stress in depression. The ways in which stress contributes to the development of MDD symptoms are diverse. Generally, stress enhances glutamate release and induces structural changes in specific areas of the brain [5] . Ketamine is considered to counterbalance these changes by inducing synaptic growth in areas that are downregulated by stress, such as the PFC and the hippo-
campus [6] . However, there are cases in which scientists have not been able to induce depression by stressing the mice, suggesting that there might be other mechanisms that make these mice resilient – just as humans experience stressful life events but do not necessarily develop MDD [12] . Understanding the underlying mechanisms of resilience can help us find new, more effective treatments with preventative effects.
One of the mechanisms identified involves BDNF released from dopamine neurons in the VTA-NAc system. Susceptible mice suffered from all kinds of depression-like symptoms, like anxiety, weight loss, and abnormalities in their circadian rhythm. Unlike their resilient counterparts, they showed a significant increase in BDNF in the NAc compared to non-stressed controls – a finding that can also be confirmed by post-mortem studies on humans with MDD [12] . Furthermore, several differences in gene expression in the VTA and NAc were identified in susceptible vs. resilient mice. Among the upregulated genes in resilient mice, some encode potassium channels in the VTA, which are expected to reduce excitability of these neurons. Scientists have hypothesized that this might counterbalance a mechanism by which dopamine neurons in the VTA get overly excited during a stressful experience. Overexpressing the same potassium channels in susceptible mice decreased firing of VTA dopamine neurons and made susceptible mice more resilient while decreasing their BDNF levels in the NAc. Hence, an increase in potassium channel activity represents a mechanism for resilience against stress-induced symptoms of depression. We can also conclude that elevated VTA activity might result in the observed increase in BDNF release into the NAc [12] .
A recent study reveals ketamine’s potential to be used as a vaccine-like preventative treatment for people with a genetic predisposition for depression [13] . The researchers tested the effects of a single dose of ketamine in different mouse models for depression and had astonishing results: a single injection of ketamine administered before stress exposure protected the mice against most behavioral hallmarks of depression, such as immobility, social avoidance, failure to escape from electric shocks (or so-called “learned helplessness”), and the already mentioned anxiety and weight loss. Despite its short half-life of 2 to 2.5 hours in rodents, the effects of ketamine lasted for at least three weeks. Apparently, ketamine can induce persistent stress resilience in a self-maintaining manner [13] . Although ketamine seems to affect the dopamine system by increasing stress-resilience, the underlying mechanism is still unidentified [9] .
Ketamine as the Prototype of Next-Generation Antidepressants
Due to its long-lasting and quickly induced effects, ketamine is potentially more effective than current antidepressants. Ketamine modifies the glutamate system, the dominant excitatory pathway in the brain, which is involved in our reaction to stress, emotional responses, and neuroplasticity. However, the drug has notable side effects that interfere with our cognition. For instance, ketamine often induces psychotic symptoms, such as hallucinations, false beliefs, and severe impairments in judgment and other cognitive processes [14] . Under its nickname “Special K,” ketamine is often sold as a street drug, adding a sour note to its potential as a treatment [1] . In addition, finding a dosage that reliably induces ketamine’s antidepressant effects but does not cross the line towards its anesthetic effects has been a major challenge in many studies [2, 7] .
The side effects, its bad reputation as an abused drug, the problem of finding the right dosage, the unspecific targeting of NMDA-Rs, and the potential of further undefined and unwanted effects will likely keep ketamine from being used outside of a well-monitored hospital setting. Nevertheless, ketamine can be considered the prototype of next-generation antidepressants that target the brain’s glutamate circuitry. Current research is trying to better understand the underlying molecular mechanisms to develop more targeted NMDA-R modulators that allow for better control and less side effects. Esketamine, a promising candidate for widespread MDD therapy, is currently in phase III trials for intravenous administration and in phase I for nasal application [15] . Esketamine is an enantiomer of ketamine, which means that its chemical structure is “mirrored” along a vertical axis. Esketamine and other drugs derived from ketamine could possibly be applied to a broad spectrum of psychiatric diseases. Ketamine has also been shown to treat bipolar disorder and post-traumatic stress disorder, and since glutamate is a major messenger acting throughout the nervous system, it is very likely that drugs targeting this system could be used to treat a variety of diseases [1, 6, 7] .
- Brachman, R. (2016). Could a Drug Prevent Depression and PTSD? TED Talk. Retrieved 2 April 2017 from https://www.ted.com/talks/rebecca_brachman_could_a_drug_prevent_depression_and_ptsd
- Dutta, A., McKie, S., & William Deakin, J.F. (2015). Ketamine and Other Potential Glutamate Antidepressants. Psychiatry Research. 225 (1-2), 1-13. doi: 10.1016/j.psychres.2014.10.028
- American Psychiatric Association. (2013). Diagnostic and statistical manual of mental disorders : DSM-5. (5th ed.). Washington, D.C.; Arlington, VA: American Psychiatric Association; American Psychiatric Association.
- Trivedi, M et al. (2006). Evaluation of Outcomes With Citalopram for Depression Using Measurement-Based Care in STARD: Implications for Clinical Practice. American Journal of Psychiatry, 163(1), 28-40. doi: 10.1176/appi.ajp.163.1.28
- Sanacora, G., Treccani, G., & Popoli, M. (2012). Towards a glutamate hypothesis of depression: An emerging frontier of neuropsychopharmacology for mood disorders: An emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology, 62 (1), 63-77. doi: 10.1016/j.neuropharm.2011.07.036
- Duman, R., Aghajanian, G., Sanacora, G., & Krystal, J. (2016). Synaptic plasticity and depression: New insights from stress and rapid-acting antidepressants. Nature Medicine, 22(3), 238-49. doi: 10.1038/nm.4050
- Mathews, D.C., Henter, I.D., & Zarate, C.A. (2012).Targeting the Glutamatergic System to Treat Major Depressive Disorder: Rationale and Progress to Date. Drugs. 72 (10) ,1313-1333. doi: 10.2165/11633130-000000000-00000
- Eagleman, D., & Downar, J (2016). Brain and Behavior. Oxford University Press, 293-294.
- Belujon, P. & Grace, A.A. (2014). Restoring Mood Balance in Depression: Ketamine Reverses Deficit in Dopamine-Dependent Synaptic Plasticity. Biological Psychiatry, 76 (12), 927-936., doi: 10.1016/j.biopsych.2014.04.014
- Krishnan, V., & Nestler, E.J. (2008). The Molecular Neurobiology of Depression. Nature. 455(7215), 894 – 902. doi: 10.1038/nature07455
- Li, N. et al. (2010). MTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. 329, 959-964. doi: 10.1126/science.1190287
- Krishnan, V. et al. (2007). Molecular Adaptations Underlying Susceptibility and Resistance to Social Defeat in Brain Reward Regions. Cell.131 (2), 391-404. doi: 10.1016/j.cell.2007.09.018
- Brachman, R. et al. (2016). Ketamine as a Prophylactic Against Stress-Induced Depressive-like Behavior. Biological Psychiatry,79 (9), 776-786. doi: 10.1016/j.biopsych.2015.04.022
- Psychosis. In Encyclopædia Britannica. Retrieved from https://www.britannica.com/science/psychosis
- Thomas, R., Cetin, M., Baker, G., & Dursun, S. (2016). Comment on FDA’s Breakthrough Therapy Designation of Intranasal Esketamine for the Treatment of Major Depressive Disorder with Imminent Risk of Suicide. Klinik Psikofarmakoloji Bulteni, 26(4), 329-331. doi: 10.5455/bcp.20161027122045