
If you asked researchers for a list of the last decade’s most important discoveries, most would include CRISPR-Cas9, a system that has the ability to find a sequence within a strand of DNA, cut it out, and replace it with a new sequence. What makes CRISPR so special is how efficient it is, making modification of the genome almost effortless. Since its discovery in 2012, CRISPR-Cas9 has become a central interest of medical research in nearly every field and won its original creators the 2020 Nobel Prize in Chemistry.
In the public eye, CRISPR-Cas9 has become synonymous with gene therapy as a whole. But even if CRISPR-Cas9 does not encompass the entirety of gene therapy as a practice, its prominence has ushered in a new paradigm where every genetic condition or disease could potentially be treated with gene therapy. Although CRISPR treatments are still experimental, the FDA has recently begun to approve gene therapies. Recent progress has been made in the treatment of an eye condition called Leber congenital amaurosis (LCA), an inherited degenerative disease that is caused by a mutation in the RPE65 protein [1]. Luxturna, a gene therapy medication approved in 2020, delivers a working copy of the RPE65 gene into the retinal cells. The cell machinery uses this delivered genetic information to produce the correct protein. While this treatment doesn’t modify the human genome, it still adequately replaces the products that dysfunctional genes fail to produce.
But for all the advances in gene therapy for conditions of the spine, eyes, or pancreas, the same excitement over CRISPR and gene therapy is less common, or at least more reserved, in the world of neuroscience. Why is this? Primarily, it boils down to the fact that the brain is really complex, more so than other organs.
Features that would make diseases of the eye or pancreas easier to treat are rare when discussing neurological conditions. Even if a neurological condition is connected to genetics, it is usually difficult to explain the disease with a single gene [2]. Because neurological diseases are often the result of multiple genes, these diseases tend to involve regions of the brain, making it difficult for scientists to find a single therapeutic target. Like other diseases, neurological conditions are degenerative in the sense that the condition worsens over time. But unlike other diseases, neurodegenerative diseases are difficult to diagnose early enough for gene therapy to still be effective. Abnormalities in fetal brain development often result in irreversible damage to the brain, rendering gene therapy futile [2]. When combined, these problems have slowed the adoption of gene therapy into the treatment of neurodevelopmental disorders.
Regardless, neuroscientists across the globe are taking small steps that bring the prospects of gene therapy closer to reality. Unexpectedly, much of this progress has taken place among a niche community of scientists researching Rett syndrome, a rare genetic neurodevelopmental disorder. The findings of these scientists challenge the assertion that neurodevelopmental disorders are irreversible and give hints about how gene therapy can be utilized to treat this class of diseases. Research into Rett Syndrome indicates that gene therapy could even treat impairments in the fully-developed brains of adults, giving new insights into the adaptability of the human brain.
What is Rett Syndrome?
Even among neuroscientists, the prominent role that Rett syndrome has played over the last decade in gene therapy has been surprising. Rett syndrome is so rare that its identification only gained general recognition among the medical community in 1983, years after research into gene therapy had already begun in the 1970s [3]. But even though Rett’s remains a niche topic of research, the body of work contributed by the few scientists studying the disease has shaped the way that scientists approach gene therapy.
Rett syndrome is a neurodevelopmental disorder often characterized as a form of autism. Rett’s primarily affects girls, occurring in less than 1 in 10,000 live female births [4]. Its symptoms manifest over several main stages [5]. Following normal development during the first 6-18 months of life, developmental progress stagnates. Between the ages of 1 and 4, patients experience rapid regression, with the loss of acquired motor and communication skills, breathing irregularities, and stagnated skull growth. Afterwards, symptoms remain relatively consistent, with further deterioration possible in the form of seizures and wheelchair dependency [5].
Progress in the understanding of the mechanisms behind Rett syndrome can be traced through the groundbreaking research of Huda Zoghbi and Adrian Bird, two scientists who have studied Rett’s for more than 30 years. In 1992, Bird was the first to discover a protein called MeCP2, which plays a role in the methylation of DNA [6]. Adding a methyl group to a strand of DNA changes its expression pattern, making MeCP2 a powerful protein that ensures gene products are produced in correct proportions. Zoghbi discovered in the same year that Rett’s is caused by a gene on the X chromosome, which explains why it primarily affects girls [7]. Then, in 1999, Zoghbi identified the MeCP2 protein as the cause of the disease [8]. With the link between Rett’s and MeCP2 established, parallel work in the Zoghbi and Bird labs led to several striking advances. Bird’s Lab not only showed that MeCP2 is particularly abundant in mature brain cells, but that it also interacts with methylated DNA and a complex which decreases the expression of DNA [9]. Zoghbi’s Lab applied this finding to Rett’s by demonstrating that the disruption of these interactions occurs in those with Rett syndrome [10]. Following this discovery, both labs developed mice models of Rett syndrome. By recreating the characteristic symptoms of MeCP2 deficiency in mice, the labs further established irregular MeCP2 expression as the cause of Rett syndrome and its symptoms.
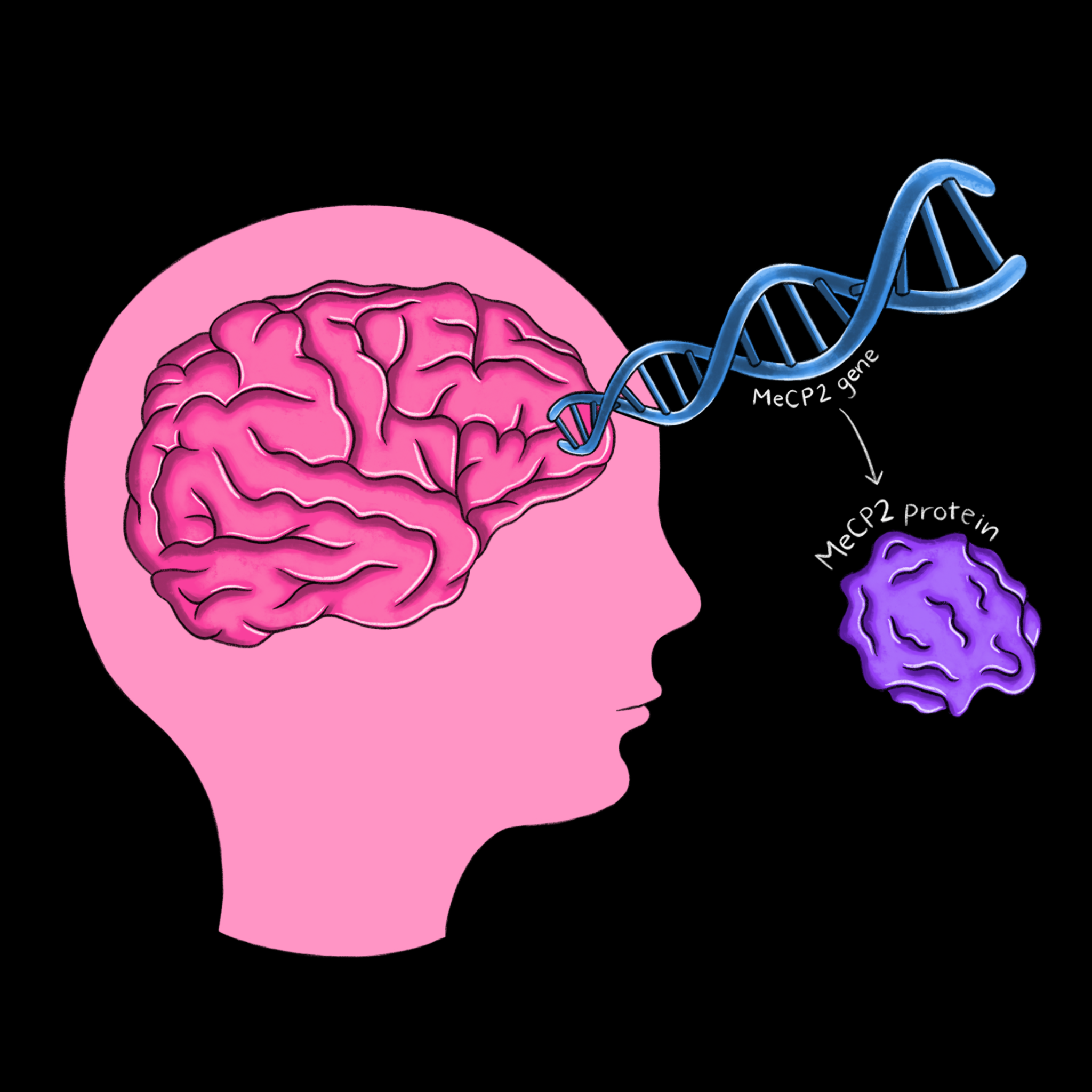
Zoghbi and Bird’s work has important implications for the treatment of neurodevelopmental disorders through gene therapy. Because Rett syndrome does not cause neuronal death, the Bird Lab investigated in 2007 whether restoration of MeCP2 protein levels could reverse the symptoms of the disease [11]. In mice genetically engineered to have the MeCP2 gene “turned off”, reactivation of the gene and production of the MeCP2 protein corrected all symptoms of the disease, including fixing the structures of neurons long after symptom onset. The Zoghbi Lab had corollary results, showing that the disease can be triggered in normal adult mice by inducing the deletion of the MeCP2 gene [12]. The introduction of MeCP2 can quickly alleviate the symptoms of Rett’s while its deletion has the immediate opposite effect of causing new symptoms to appear.
The astonishing results of the Zoghbi and Bird Lab push back against long-held assumptions about the permanence of neurodevelopmental disorders. So distinct were the features of Rett’s from existing neurodevelopmental disorders that the researchers in the Bird Lab were hesitant to consider the disease as neurodevelopmental. But in the years following these discoveries, researchers have reinterpreted Rett’s as part of a new category of treatable neurodevelopmental disorders. While the molecular and cellular processes behind Rett syndrome are still not fully understood, the disease remains remarkably simple in that it is caused by only one gene. If gene therapy targeting this particular sequence can successfully treat Rett syndrome, the technique bodes well for the treatment of other diseases caused by more complex arrays of genes.
Gene Therapy Techniques
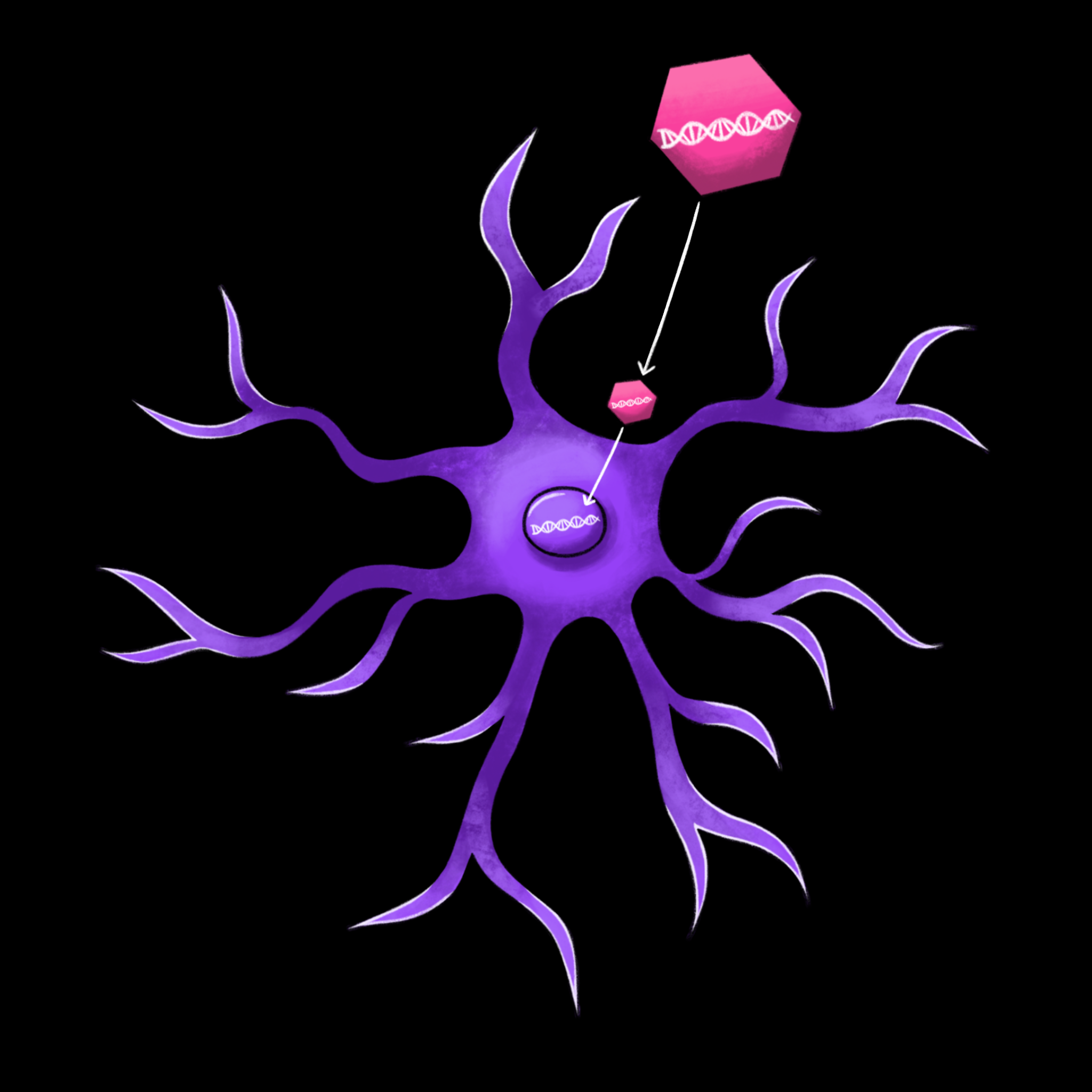
Even if researchers demonstrate that gene therapy can effectively treat Rett syndrome in mice, there are still many steps before the development of treatments for humans. Gene therapy is largely accomplished through viral vectors, taking advantage of the cell-infiltrating properties of viruses to deliver new DNA into the human cell. Viruses reproduce by delivering viral DNA into the nucleus of a cell, where the machinery of the cell uses the DNA to create new copies of the virus. In the human body, viruses are almost always harmful. But in gene therapy, scientists replace the viral DNA within the shell of the virus with synthesized DNA. Instead of producing new viruses, the cell uses the synthesized DNA to produce helpful protein products.
With the use of viral vectors, however, comes intrinsic risks and complications. The first of these complications is the immune system, which is designed to identify and eliminate viruses. [13]. The immune response can destroy cells which contain the virus and the synthesized genetic material within it, rendering the entire treatment ineffective. Additionally, the immune response can cause severe side effects, such as inflammation and, in severe cases, organ failure [13].
Viruses are also tricky to engineer in a way that provides both safe and effective gene therapy. Because viruses can affect more than one type of cell, the altered viruses may infect additional cells beyond the ones targeted [13]. If scientists are unable to specifically target certain cells, there may be unintended effects in other cells. The chances of a strong immune response go up, increasing the risks of serious inflammatory side effects. In addition, if viruses replicate uncontrollably in the body, they can cause disease and, again, an immune response. Thus, scientists must ensure that the viruses propagate enough to deliver the desired gene, but without causing infection. Finally, scientists must be careful about how the newly introduced genetic material interacts with the existing genome. If this genetic material integrates into the genome improperly, its effects on gene expression can cause cancer [13]. Effective gene therapy minimizes the risk of these side effects while still delivering enough genetic material for the adequate, long-term production of a desired product.
In preliminary studies using gene therapy to treat Rett syndrome, researchers have primarily used the adeno-associated virus (AAV) vector. AAVs are small viruses which do not replicate and elicit a minimal immune response, eliminating many of the complications associated with conventional viral vectors [13]. Compared to other forms of gene therapy, AAVs are useful because they work on multiple cell types in the central nervous system (CNS), including neurons, astrocytes, and glial cells [14]. The typical drawback of AAVs is that they do not integrate the genetic material into the host genome, instead leaving the DNA inside the nucleus where it can be expressed [15]. During cell division, the AAV is not replicated, so when a cell divides only one of the daughter cells will have the added gene. Over time, this results in the expression of the gene in fewer target cells. As the expression of the gene reduces, the treatment becomes less effective. This drawback is not an issue for treatments that target the brain because neurons and glial cells are terminally differentiated, meaning that they persist over the entire human lifespan without dividing. Thus, only a single administration of the drug is necessary for long-term treatment. These features make AAVs the main focus of research in not only Rett syndrome, but also other treatments that target CNS diseases.
Several recent advances have brought the dream of successful gene therapy for Rett syndrome closer to reality. In 2004, Zoghbi and her associates discovered in mice that, in addition to underexpression, MeCP2 overexpression resulted in neurodevelopmental disorders similar to Rett syndrome [16]. This finding was followed up in 2005 by the discovery in humans of a rare disease caused by MeCP2 overexpression, later coined MeCP2 duplication syndrome, which has similar symptoms as Rett’s [17]. These findings mean that scientists can’t cure Rett syndrome by simply delivering the MeCP2 gene to as many cells as possible. If they do, patients will begin exhibiting the symptoms of MeCP2 duplication syndrome, which itself is a neurodevelopmental disorder. In order to effectively treat Rett’s, researchers must find the cells that contribute most to the disease, then design AAV vectors that target those specific cells. Researchers in the Zoghbi group have already begun this process. In a 2016 mice study, they found that restoring MeCP2 in only inhibitory neurons resulted in the reduction of disease symptoms [18]. While parallel studies have shown that excitatory neurons still play a role in the disease, this finding is an important first step in the identification of target neurons for future therapeutics [19].
Even if scientists eventually identify the subset of neurons most responsible for Rett’s, targeting these neurons requires advances in AAV technology. Luckily, as research into AAVs has continued, scientists have begun to develop better methods to target AAVs to specific cell types and neurons. Gene therapy using AAVs relies on the unique capsid of the virus, the protein shell which interacts with cell membranes and facilitates infiltration of the cell. Scientists use a range of about ten different capsids depending broadly on which cells they want to target [20]. But in more complex cases like Rett’s, these capsids don’t target the desired cells with enough specificity. As a result, scientists have begun engineering capsids that contain desired attributes. When capsids are modified, they gain or lose the ability to interact with specific receptors, thus altering the cell types that they target. The two main methods for engineering capsids are the use of hybrid capsids and short peptides [21]. Hybrid capsids are assembled from pieces of naturally occurring capsids, resulting in a capsid that can better target the surface of a cell. As techniques for the chemical modification of the viral capsids improve, scientists have also begun experimenting with small protein chains called short peptides. By inserting these peptides into specific locations within the capsid, scientists can facilitate interactions with specific target cell receptors. When combined, these two strategies have the potential to greatly improve the specificity of gene therapy in the brain.
Further Studies
While therapeutics for Rett syndrome in humans are still far off, the pioneering work by Zoghbi, Bird, and other researchers on the disease has already provided new insights on the study of other diseases. When the Bird group discovered that Rett’s was reversible in mice, the implications of the study challenged assumptions regarding neurodevelopmental disorders. Most neurodevelopmental disorders are not reversible in adulthood due to permanent structural deformations that occur over years of development. This problem is compounded by the fact that as the brain matures, it loses its plasticity, the ability to rearrange and create new connections between neurons. The discovery of Rett syndrome’s reversibility alters this paradigm. While researchers still view the treatment of most neurodevelopmental disorders as non-curative, the unique attributes of Rett syndrome indicate that there may be a distinct subset of related neurodevelopmental disorders that are treatable.
After the reversibility of Rett’s was discovered, researchers began to group it with other diseases, both by reinterpreting past results and going forward with new studies. Among these diseases are neurofibromatosis, tuberous sclerosis, fragile X syndrome, and Angelman syndrome [22]. Inspired by the demonstrated reversibility of Rett’s, research groups have shown in mice models that these diseases are potentially reversible after the onset of symptoms [23][24][25]. Like with Rett’s, these diseases are less the result of structural problems and are more due to faulty connections between neurons. As researchers further elucidate the neural mechanisms behind Rett’s, the common characteristics of this group of diseases becomes better understood. Rett’s is now understood to be caused, in part, by a global imbalance in excitatory and inhibitory signaling. The proportion of excitatory to inhibitory signaling determines the frequency at which neurons fire, making the excitation-inhibition balance important for normal brain function. Excitation-inhibition balance problems often result in seizures, making it a characteristic symptom of this group of neurodevelopmental diseases. The excitation-inhibition balance observed in Rett syndrome has several implications for other related diseases. First, it has been hypothesized that increased levels of inhibition suppress plasticity during key periods of brain development [26]. As a result, treatments that decrease inhibition have the opportunity to restore this plasticity and reopen developmental windows in the adult brain. The successful treatment of Rett’s in adult mice backs up this hypothesis, improving the outlook for the treatment of other excitation-inhibition-related diseases. Second, the importance of excitation-inhibition in Rett’s connects the disease to autism spectrum disorder and Down syndrome, two common neurodevelopmental disorders with similar symptoms that also involve excitation-inhibition [27]. Although these diseases are more complex than Rett’s, scientists can apply their findings from models of Rett’s and related genetic disorders to broader neurodevelopmental diseases. If, for example, Rett’s researchers narrow down their target neurons for gene therapy and develop specific AAVs, these findings would be invaluable for the study of other excitation-inhibition diseases that involve similar groups of neurons.
Conclusion
Regardless of how promising gene therapy appears to be for the treatment of neurodevelopmental disorders like Rett’s, scientists are wary of predicting when these therapies will become available for humans. Gene therapy has a long history of failure, even in simple diseases located in less complicated organs. Still, the past 30 years of research on Rett’s is a testament to how quickly unexpected discoveries can change the direction of the field of neuroscience. The Zoghbi and Bird labs, in addition to a host of new researchers, continue to diligently work on understanding the mechanisms of the disease. Rapid technological advances mean that scientists are now able to follow up on previous studies with greater efficiency and better precision. CRISPR in particular has the potential to simplify basic research and eliminate some of the limitations of gene therapy. So while CRISPR is not the embodiment of gene therapy, it is an important tool with the ability to accelerate progress on Rett’s and other diseases in the coming years.
Rett’s will always be a niche genetic disease, so rare that few in the general population will have heard of it. Still, the unique attributes of Rett’s makes it extremely valuable as a subject of research interest. As scientists begin to understand how to treat Rett’s, these findings will inevitably be applied to diseases like autism, dementia, and Parkinson’s.
References
- Pierce, E. A., & Bennett, J. (2015). The Status of RPE65 Gene Therapy Trials: Safety and Efficacy. Cold Spring Harbor Perspectives in Medicine, 5(9), a017285. https://doi.org/10.1101/cshperspect.a017285
- Martier, R., & Konstantinova, P. (2020). Gene Therapy for Neurodegenerative Diseases: Slowing Down the Ticking Clock. Frontiers in Neuroscience, 14, 580179. https://doi.org/10.3389/fnins.2020.580179
- Kyle, S. M., Vashi, N., & Justice, M. J. (2018). Rett syndrome: a neurological disorder with metabolic components. Open Biology, 8(2), 170216. https://doi.org/10.1098/rsob.170216
- Burd, L., Randall, T., Martsolf, J. T., & Kerbeshian, J. (1991). Rett syndrome symptomatology of institutionalized adults with mental retardation: comparison of males and females. American Journal of Mental Retardation, 95(5), 596–601.
- Neul, J. L., Kaufmann, W. E., Glaze, D. G., Christodoulou, J., Clarke, A. J., Bahi-Buisson, N., Leonard, H., Bailey, M. E., Schanen, N. C., Zappella, M., Renieri, A., Huppke, P., Percy, A. K., & RettSearch Consortium (2010). Rett syndrome: revised diagnostic criteria and nomenclature. Annals of Neurology, 68(6), 944–950. https://doi.org/10.1002/ana.22124
- Lewis, J. D., Meehan, R. R., Henzel, W. J., Maurer-Fogy, I., Jeppesen, P., Klein, F., & Bird, A. (1992). Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell, 69(6), 905–914. https://doi.org/10.1016/0092-8674(92)90610-o
- Ellison, K. A., Fill, C. P., Terwilliger, J., DeGennaro, L. J., Martin-Gallardo, A., Anvret, M., Percy, A. K., Ott, J., & Zoghbi, H. (1992). Examination of X chromosome markers in Rett syndrome: exclusion mapping with a novel variation on multilocus linkage analysis. American Journal of Human Genetics, 50(2), 278–287.
- Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., & Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nature Genetics, 23(2), 185–188. https://doi.org/10.1038/13810
- Lyst, M. J., Ekiert, R., Ebert, D. H., Merusi, C., Nowak, J., Selfridge, J., Guy, J., Kastan, N. R., Robinson, N. D., de Lima Alves, F., Rappsilber, J., Greenberg, M. E., & Bird, A. (2013). Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nature Neuroscience, 16(7), 898–902. https://doi.org/10.1038/nn.3434
- Samaco, R. C., Mandel-Brehm, C., Chao, H. T., Ward, C. S., Fyffe-Maricich, S. L., Ren, J., Hyland, K., Thaller, C., Maricich, S. M., Humphreys, P., Greer, J. J., Percy, A., Glaze, D. G., Zoghbi, H. Y., & Neul, J. L. (2009). Loss of MeCP2 in aminergic neurons causes cell-autonomous defects in neurotransmitter synthesis and specific behavioral abnormalities. Proceedings of the National Academy of Sciences of the United States of America, 106(51), 21966–21971. https://doi.org/10.1073/pnas.0912257106
- Guy, J., Gan, J., Selfridge, J., Cobb, S., & Bird, A. (2007). Reversal of neurological defects in a mouse model of Rett syndrome. Science, 315(5815), 1143–1147. https://doi.org/10.1126/science.1138389
- Ben-Shachar, S., Chahrour, M., Thaller, C., Shaw, C. A., & Zoghbi, H. Y. (2009). Mouse models of MeCP2 disorders share gene expression changes in the cerebellum and hypothalamus. Human Molecular Genetics, 18(13), 2431–2442. https://doi.org/10.1093/hmg/ddp181
- Lentz, T. B., Gray, S. J., & Samulski, R. J. (2012). Viral vectors for gene delivery to the central nervous system. Neurobiology of Disease, 48(2), 179–188. https://doi.org/10.1016/j.nbd.2011.09.014
- Van der Perren, A., Toelen, J., Carlon, M., Van den Haute, C., Coun, F., Heeman, B., Reumers, V., Vandenberghe, L. H., Wilson, J. M., Debyser, Z., & Baekelandt, V. (2011). Efficient and stable transduction of dopaminergic neurons in rat substantia nigra by rAAV 2/1, 2/2, 2/5, 2/6.2, 2/7, 2/8 and 2/9. Gene Therapy, 18(5), 517–527. https://doi.org/10.1038/gt.2010.179
- Naso, M. F., Tomkowicz, B., Perry, W. L., 3rd, & Strohl, W. R. (2017). Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs, 31(4), 317–334. https://doi.org/10.1007/s40259-017-0234-5
- Collins, A. L., Levenson, J. M., Vilaythong, A. P., Richman, R., Armstrong, D. L., Noebels, J. L., David Sweatt, J., & Zoghbi, H. Y. (2004). Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Human Molecular Genetics, 13(21), 2679–2689. https://doi.org/10.1093/hmg/ddh282
- Ta, D., Downs, J., Baynam, G., Wilson, A., Richmond, P., & Leonard, H. (2022). A brief history of MECP2 duplication syndrome: 20-years of clinical understanding. Orphanet Journal of Rare Diseases, 17(1), 131. https://doi.org/10.1186/s13023-022-02278-w
- Ure, K., Lu, H., Wang, W., Ito-Ishida, A., Wu, Z., He, L. J., Sztainberg, Y., Chen, W., Tang, J., & Zoghbi, H. Y. (2016). Restoration of Mecp2 expression in GABAergic neurons is sufficient to rescue multiple disease features in a mouse model of Rett syndrome. eLife, 5, e14198. https://doi.org/10.7554/eLife.14198
- Meng, X., Wang, W., Lu, H., He, L. J., Chen, W., Chao, E. S., Fiorotto, M. L., Tang, B., Herrera, J. A., Seymour, M. L., Neul, J. L., Pereira, F. A., Tang, J., Xue, M., & Zoghbi, H. Y. (2016). Manipulations of MeCP2 in glutamatergic neurons highlight their contributions to Rett and other neurological disorders. eLife, 5, e14199. https://doi.org/10.7554/eLife.14199
- Castle, M. J., Turunen, H. T., Vandenberghe, L. H., & Wolfe, J. H. (2016). Controlling AAV Tropism in the Nervous System with Natural and Engineered Capsids. Methods in Molecular Biology, 1382, 133–149. https://doi.org/10.1007/978-1-4939-3271-9_10
- O'Carroll, S. J., Cook, W. H., & Young, D. (2021). AAV Targeting of Glial Cell Types in the Central and Peripheral Nervous System and Relevance to Human Gene Therapy. Frontiers in Molecular Neuroscience, 13, 618020. https://doi.org/10.3389/fnmol.2020.618020
- Ehninger, D., Li, W., Fox, K., Stryker, M. P., & Silva, A. J. (2008). Reversing neurodevelopmental disorders in adults. Neuron, 60(6), 950–960. https://doi.org/10.1016/j.neuron.2008.12.007
- Dölen, G., Osterweil, E., Rao, B. S., Smith, G. B., Auerbach, B. D., Chattarji, S., & Bear, M. F. (2007). Correction of fragile X syndrome in mice. Neuron, 56(6), 955–962. https://doi.org/10.1016/j.neuron.2007.12.001
- Bourtchouladze, R., Lidge, R., Catapano, R., Stanley, J., Gossweiler, S., Romashko, D., Scott, R., & Tully, T. (2003). A mouse model of Rubinstein-Taybi syndrome: defective long-term memory is ameliorated by inhibitors of phosphodiesterase 4. Proceedings of the National Academy of Sciences of the United States of America, 100(18), 10518–10522. https://doi.org/10.1073/pnas.1834280100
- Meikle, L., Pollizzi, K., Egnor, A., Kramvis, I., Lane, H., Sahin, M., & Kwiatkowski, D. J. (2008). Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. The Journal of Neuroscience, 28(21), 5422–5432. https://doi.org/10.1523/JNEUROSCI.0955-08.2008
- Spolidoro, M., Sale, A., Berardi, N., & Maffei, L. (2009). Plasticity in the adult brain: lessons from the visual system. Experimental Brain Research, 192(3), 335–341. https://doi.org/10.1007/s00221-008-1509-3
- Lintas, C., Sacco, R., & Persico, A. M. (2012). Genome-wide expression studies in autism spectrum disorder, Rett syndrome, and Down syndrome. Neurobiology of Disease, 45(1), 57–68. https://doi.org/10.1016/j.nbd.2010.11.010